|
Nomenclature of Carboxylic Acid Derivatives
|
Acyl
Chlorides
Functional group suffix = oyl chloride
(review) |
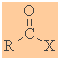 |
|
Anhydrides
Functional group suffix = alkanoic anhydride
(review) |
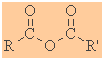 |
|
Thioesters
Functional group suffix = alkyl -oate
Functional group prefix = alkoxycarbonyl- or carbalkoxy- |
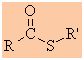 |
|
Esters
Functional group suffix = alkyl -oate
(review)
Functional group prefix = alkoxycarbonyl- or carbalkoxy-
Cyclic esters are called lactones |
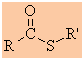 |
|
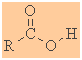
Carboxylic Acids
Functional group suffix = -oic acid (review)
Functional group prefix = carboxy- |
 |
|
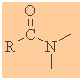
Amides
Functional group suffix = amide
(review)
Functional group prefix = carbamoyl-
Cyclic amides are called lactams |
 |
|
Nitriles
Functional group suffix = nitrile or -onitrile
(review)
Functional group prefix = cyano- |
 |
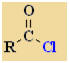
Acyl Halides or
Acid Halides
|
Nomenclature |
Formula |
|
Functional class name = acyl or acid halide
Substituent suffix = -oyl halide |
|
-
Acyl or acid
halides are derivatives of carboxylic acids.
-
The root name
is based on the longest chain including the carbonyl group of the acyl
group.
-
Since the
acyl group is at the end of the chain, the C=O carbon must be C1.
-
The acyl
halide suffix is appended after the hydrocarbon suffix minus the "e" :
e.g. -ane + -oyl halide = -anoyl halide etc.
-
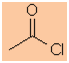
The most
common halide encountered is the chloride, hence acyl or acid chlorides,
e.g. ethanoyl chlorid
-
Functional group is an acyl halide therefore suffix = -oyl
chloride
-
Hydrocarbon structure is an alkane therefore -an-
-
The longest continuous chain is C2 therefore root = eth
ethanoyl chloride |
CH3C(=O)Cl |
-
Functional group is an acyl halide therefore suffix = -oyl
chloride
-
Hydrocarbon structure is an alkane therefore -an-
-
The longest continuous chain is C4 therefore root = but
butanoyl chloride |
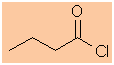
CH3CH2CH2C(=O)Cl |
-
Functional group is an acyl halide therefore suffix = -oyl
chloride
-
Hydrocarbon structure is an alkane therefore -an-
-
The longest continuous chain is C3 therefore root = prop
-
The branch is a C1 alkyl group i.e. a methyl group
-
The functional groups requires numbering from the right
as drawn, the substituent locant is 2-
2-methylpropanoyl chloride |
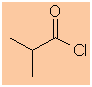
(CH3)2CHC(=O)Cl |
Acid Anhydrides
|
Nomenclature |
Formula |
|
Functional class name = alkanoic anhydride
Substituent suffix = -oic anhydride |
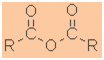 |
-
As the name
implies, acid anyhydrides are derivatives of carboxylic acids.
-
In principle,
they can be symmetric (where the two R groups are identical) or asymmetric
(where the two R groups are different).
-
Symmetric
anhydrides are the most common, they are named as alkanoic anhydrides
-
Asymmetric
anhydrides are name in a similar fashion listing the alkyl groups in
alphabetical order.
-
Cyclic
anhydrides derived from dicarboxylic acids are name as -dioic anhydrides.
-
Functional group is an acid anhydride therefore suffix = -oic
anhydride
-
Hydrocarbon structure is an alkane therefore -an-
-
The longest continuous chain is C2 therefore root = eth
ethanoic anhydride |
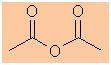
CH3C(=O)OC(=O)CH3 |
-
Functional group is an acid anhydride therefore suffix = -oic
anhydride
-
Hydrocarbon structure is an alkane therefore -an-
-
The longest continuous chain is C4 therefore root = but
-
The other group is C3 = prop
butanoic propanoic anhydride |
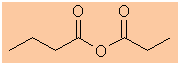 |
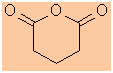
-
Functional group is a cyclic acid anhydride therefore suffix = -dioic
anhydride
-
Hydrocarbon structure is an alkane therefore -an-
-
The longest continuous chain is C5 therefore root = pent
pentandioic anhydride |
 |
Back
to the top
Esters
|
Nomenclature |
Formula |
|
Functional class name = alkyl alkanoate
Substituent suffix = -oate |
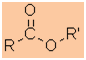 |
-
Esters are
alkyl derivatives of carboxylic acids.
-
The easiest
way to deal with naming esters is to recognise the carboxylic acid and the
alcohol that they can be prepared from.
-
The general
ester, RCO2R' can be derived from the carboxylic acid RCO2H
and the alcohol HOR'
-
The first
component of an ester name, the alkyl is derived from the
alcohol,
R'OH.
-
The second
component of an ester name, the -oate is derived from the
carboxylic
acid,
RCO2H.
-
Alcohol
component
-
the root
name is based on the longest chain containing the -OH group.
-
The chain
is numbered so as to give the -OH the lowest possible number.
-
Carboxylic
acid
component
-
the root
name is based on the longest chain including the carbonyl group.
-
Since the
carboxylic acid group is at the end of the chain, it must be C1.
-
The ester
suffix for the acid component is appended after the hydrocarbon suffix
minus the "e" : e.g. -ane + -oate = -anoate etc.
-
The complete
ester name is the alkyl alkanoate
|
-
Functional group is an ester
-
The alcohol component here is methanol, so the alkyl = methyl
-
The acid component here is propanoic acid, so propanoate
methyl propanoate |
CH3CH2C(=O)OCH3 |
Back
to the top
Amides
|
Nomenclature |
Formula |
|
Functional class name = alkyl alkanamide
Substituent suffix = -amide |
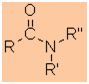 |
-
Amides are
amine derivatives of carboxylic acids.
-
The root name
is based on the longest chain including the carbonyl group of the amide
group.
-
Since the
amide group is at the end of the chain, the C=O carbon must be C1.
-
The amide
suffix is appended after the hydrocarbon suffix minus the "e" : e.g.
-ane + -amide = -anamide etc.
-
If the amide
nitrogen is substituted, the these substituents are given N- as the locant.
-
The N- locant
is listed first.
-
Functional group is an amide therefore suffix = -amide
-
Hydrocarbon structure is an alkane therefore -an-
-
The longest continuous chain is C4 therefore root = but
butanamide |
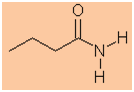
CH3CH2CH2C(=O)NH2 |
-
Functional group is an amide therefore suffix = -amide
-
Hydrocarbon structure is an alkane therefore -ane
-
The longest continuous chain is C4 therefore root = but
-
The nitrogen substituent is C1 i.e. an N-methyl
group
N-methylbutanamide |
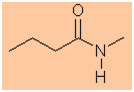
CH3CH2CH2C(=O)N(CH3)H |
-
Functional group is an amide therefore suffix = -amide
-
Hydrocarbon structure is an alkane therefore -ane
-
The longest continuous chain is C2 therefore root = eth
-
The two nitrogen substituents are C1 i.e. an N-methyl
group
-
There are two methyl groups, therefore multiplier = di-
N,N-dimethylethanamide |
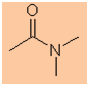
CH3C(=O)N(CH3)2 |
Back
to the top
Nitriles
|
Nomenclature |
Formula |
|
Functional class = alkyl cyanide
Functional group suffix = nitrile or -onitrile
Substituent prefix = cyano-
Notes :
-
The
cyano prefix is used in a very similar manner to haloalkanes.
-
The
cyano nomenclature is most common when the alkyl group is
simple.
-
The
nitrile suffix is used in a very similar manner to
carboxylic acids.
|
|
Cyano substituent style:
-
The root name
is based on the longest chain with the -C≡N as a substituent.
-
This root
give the alkane part of the name.
-
The chain is
numbered so as to give the -C≡N group the lowest possible locant number
Nitrile style:
-
The root name
is based on the longest chain including the carbon of the nitrile
group.
-
This root
give the alkyl part of the name.
-
Since the
nitrile must be at the end of the chain, it must be C1 and no locant needs
to be specified.
-
Nitriles can
also be named by replacing the -oic acid suffix of the corresponding
carboxylic acid with -onitrile.
|
Cyano substituent style:
-
Functional group is an alkane, therefore suffix = -ane
-
The longest continuous chain is C3 therefore root = prop
-
The substituent is a -CN therefore prefix = cyano
-
The first point of difference rule requires numbering from the
right as drawn, the substituent locant is 1-
1-cyanopropane
Nitrile style:
-
Functional group is a -C≡N, therefore suffix = -nitrile
-
Hydrocarbon structure is an alkane therefore -ane
-
The longest continuous chain is C4 therefore root = but
butanenitrile
|

CH3CH2CH2C≡N |
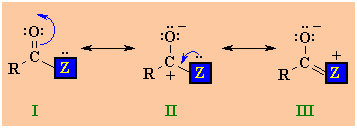
Structure of
Carboxylic Acid Derivatives
-
The carbonyl
group consists of an O atom bonded to a C atom via a double
bond in a planar, sp2 hybridisation model similar to that of a
ketone or an alkene.
-
The
heteroatom group is connected to this C=O unit via a s bond.
-
To see these
features, compare the JMOL images to the below.
-
JMOL images
of the other carboxylic acid derivatives can be found on the
previous page.
-
The resonance
interaction of the carbonyl C=O with the lone pair of the adjacent
heteroatom (structure III) has important implications on the
reactivity
-
It
also has implications for structure... Look at the JMOL image of the
amide to the right.
-
Amines and ammonia are usually pyramidal .
-
The
planar sp2 N system allows the N lone pair to align with
the C=O
p
system (see image below, with the other bonds omitted for clarity)
-
The
resonance interaction in the amide results in the C-N bond
having some double bond character (shorter, restricted rotation)
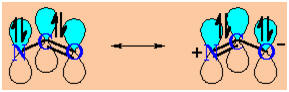 |
-
Nitriles
are
slightly different to the other derivatives in that they involved a
triple bond.
-
Nitriles consists of a N atom bonded to a C atom via a
triple bond in a linear, sp hybridisation model similar to that of
an alkyne. Compare the JMOL images to the right.
|
|
|
Reactivity of
Carboxylic Acid Derivatives
Carboxylic acid
derivatives react tend to react via nucleophilic acyl substitution where
the group on the acyl unit, R-C=O undergoes substitution:
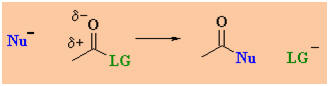
The observed reactivity order is shown below:
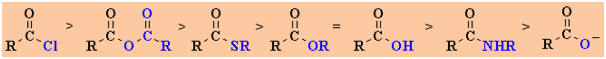
This
reactivity order is important. You should be able to understand, rationalise and
use it.
|
It is
useful to view the carboxylic acid derivatives as an acyl group,
R-C=O, with a different
substituent attached.
The important features of the carboxylic acid derivatives that
influence their reactivity are governed by this
substituent in the
following ways:
-
the effect the substituent has on the electrophilicity of the
carbonyl C .
-
if the substituent is electron donating, then the
electrophilicity is reduced, \ less reactive
-
if the substituent is electron withdrawing, the the
electrophilicity is increased, \more reactive
-
the ability of the substitutent to function as a leaving group.
|
 |
Back
to the top
There are 3
resonance structures to consider for carboxylic acid derivatives.
|
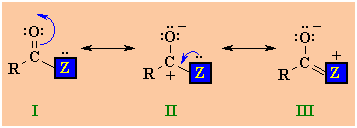 |
I
and II are similar to those of
aldehydes and ketones,
but there is also a third possibility III where a lone pair
on the heteroatom Z is able to donate electrons to the
adjacent positive center. The stronger this electron donation from
Z the less positive the carbonyl C and the less
electrophilic the carbonyl group. The ability of Z to donate
electrons is linked to its electronegativity...the more
electronegative Z is, the less the stabilising effect. |
Use the following
series of electrostatic potential maps to look at the electrophilicity of the
carbonyl C in a example of each the more common carboxylic acid
derivatives. Note how the blue colour
gradually reduces in intensity down the series.
|
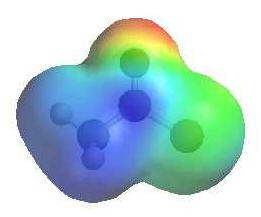 |
The
image shows the electrostatic potential for ethanoyl chloride.
The more red an area is, the
higher the electron density and the more
blue an area is, the lower
the electron density. |
|
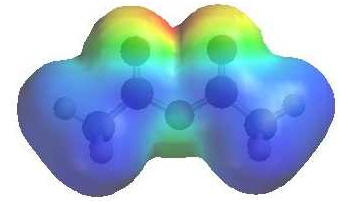 |
The
image shows the electrostatic potential for ethanoic anhydride.
The more red an area is, the
higher the electron density and the more
blue an area is, the lower
the electron density. |
|
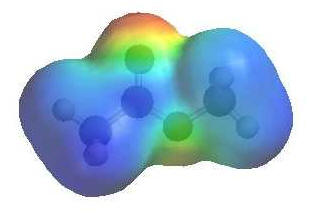 |
The
image shows the electrostatic potential for methyl ethanoate.
The more red an area is, the
higher the electron density and the more
blue an area is, the lower
the electron density. |
|
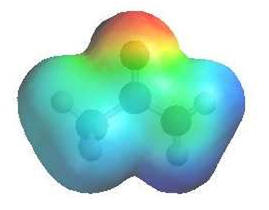 |
The
image shows the electrostatic potential for ethanamide.
The more red an area is, the
higher the electron density and the more
blue an area is, the lower
the electron density. |
|
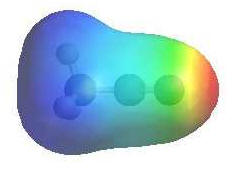 |
The
image shows the electrostatic potential for acetonitrile.
The more red an area is, the
higher the electron density and the more
blue an area is, the lower
the electron density. |
|
Derivative |
Substituent |
Electronic Effect
|
Leaving Group Ability |
Relative Reactivity |
|
Acyl
chloride |
-Cl |
withdrawing group (inductive) |
very
good |
1
(most) |
|
Anhydride |
-OC=OR |
weakly donating |
good
|
2
|
|
Thioester |
-SR |
donating |
moderate |
3
|
|
Ester |
-OR |
strongly donating |
poor
|
=4
|
|
Acid |
-OH |
strongly donating |
poor
|
=4
|
|
Amide |
-NH2,
-NR2 |
very
strongly donating |
very
poor |
5
|
|
Carboxylate |
-O- |
very,
very strongly donating |
appalling ! |
6
(least) |
It is also
useful to appreciate where aldehydes and ketones fit into the reactivity scale
towards nucleophiles:
acyl halides >
anhydrides > aldehydes > ketones > esters = carboxylic acids > amides
Overview of
Nucleophilic Acyl Substitution
Overall nucleophilic acyl
substitution is most simply represented as follows:
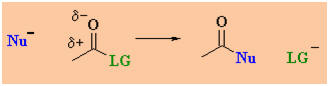
What does the
term "nucleophilic acyl substitution" imply ?
A nucleophile is an
electron rich species that will react with an electron poor species (Nu
in scheme).
An acyl group is R-C=O (where R can be alkyl or aryl)....
note the acyl group in both the starting material and the product.
A substitution note that the leaving group (LG)
is replaced by the nucleophile (Nu).
There are two fundamental events
in a nucleophilic acyl substitution reaction:
-
formation of the new s bond to
the nucleophile, Nu.
-
breaking of the s bond to the
leaving group, LG.
|
Overall, these events are the same as those in a simple nucleophilic
substitution (chapter
8), note the
fundamental similarity in the two general processes. |
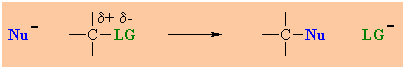
|
The difference in nucleophilic
acyl substitution is that when the nucleophile adds to the
electrophilic C, it becomes tetrahedral and an intermediate
forms, then the leaving group departs as shown below:
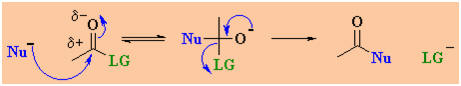
Back
to the top
Reactions for
Interconverting Carboxylic Acids Derivatives
Here is a table
that summarises the methods for interconverting carboxylicacid derivatives. The
more important reactions in emphasised in bold,and the reactions of the
parent carboxylic acids in
blue.
|
To
make ->
From
|
 |
|
 |
-
|
RCO2-
|
R'OH
|
H2O
|
R2NH
|
H2O,
HO-
|
|
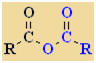 |
-
|
-
|
R'OH
|
H2O
|
R2NH
|
H2O,
HO- |
|
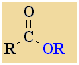 |
-
|
-
|
R'OH,
heat
H+ or B- |
H2O,
H3O+
|
R2NH
|
H2O,
HO-
heat |
|
 |
SOCl2
or PCl3 |
Heat, -H2O
|
R'OH, heat
H+ |
-
|
R2NH
heat |
HO-
|
|
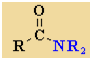 |
-
|
-
|
-
|
H2O,
H3O+
heat |
-
|
H2O,
HO-
heat |
|
 |
-
|
RCOCl
|
R'-Br
or -I |
H3O+
|
-
|
-
|
Reactions
of Carboxylic Acid Derivatives
Interconversion Reactions of Acyl Chlorides
|
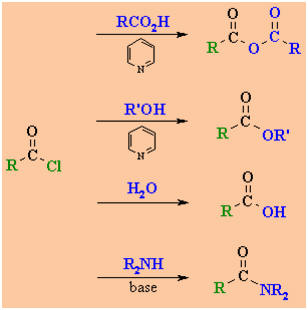
|
acid anhydrides
esters
acids
amides
|
Reaction type: Nucleophilic
Acyl Substitution
Summary
-
Acyl chlorides are the most
reactive of the carboxylic acid derivativesand therefore can be readily
converted into all other carboxylic acid derivatives(see above).
-
They are sufficiently reactive
that they react quite readily with coldwater and hydrolyse to the carboxylic
acid.
-
The HCl by-product is usually
removed by adding a base such as pyridine,C6H5N, or
triethyl amine, Et3N.
Interconversion
Reactions of Acid Anhydrides
|
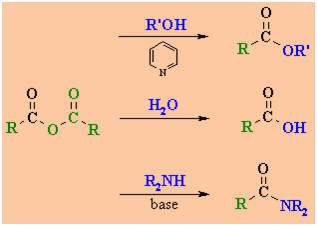 |
esters
acids
amides
|
Reaction type: Nucleophilic
Acyl Substitution
Summary
-
Acid anhydrides are the second
most reactive of the carboxylic acid derivatives and can therefore, be
fairly readily converted into the other less reactive carboxylic acid
derivatives (see above).
-
A base in often added to
neutralise the carboxylic acid by product that is formed.
Back
to the top
Interconversion
Reactions of Esters
|
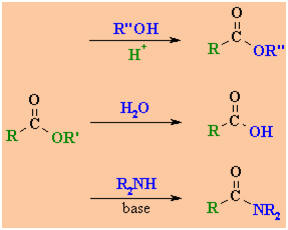 |
esters
acids
amides |
Reaction type: Nucleophilic
Acyl Substitution
Summary
-
Esters can be converted into
other esters (transesterification), the parent carboxylic acid (hydrolysis)
or amides (see above).
-
Transesterification
: heat with alcohol and acid catalyst
-
Hydrolysis: heat with aq. acid o base (e.g. aq. H2SO4
or aq. NaOH) .
-
Amide preparation
: heat with the amine, methyl or ethyl esters are the most reactive
Interconversion
Reactions of Amides
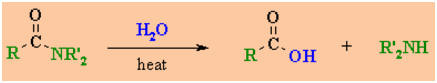
Reaction
type: Nucleophilic Acyl Substitution
Summary
-
Amides are the least reactive of
the neutral carboxylic acid derivatives.
-
The only interconversion
reaction that amides undergo is hydrolysis back to the parent carboxylic
acid and the amine.
-
Reagents : Strong acid (e.g.
H2SO4) or strong base (e.g. NaOH) / heat.
-
More details on the following
page.
Reactions
of Nitriles
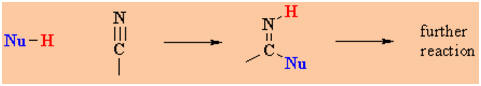
Reaction
type: Nucleophilic Addition
Overview
-
Nitriles typically undergo nucleophilic addition
to give products that often undergo a further reaction.
-
The chemistry of the nitrile
functional group, C≡N, is very similar to that of the carbonyl,
C=O of aldehydes and ketones. Compare the two schemes:
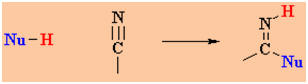 versus
versus 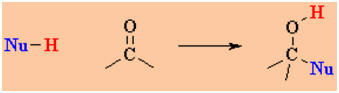
-
However, it is convenient to
describe nitriles as carboxylic acid derivatives because:
-
the oxidation state of the C
is the same as that of the carboxylic acid derivatives.
-
hydrolysis produces the
carboxylic acid
-
Like the carbonyl containing
compounds, nitriles react with nucleophiles via two scenarios:
-
Strong nucleophiles
(anionic) add directly to the C≡N to form an intermediate imine salt
that protonates (and often reacts further) on work-up with dilute acid.
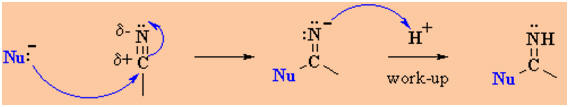
Examples of such
nucleophilic systems are : RMgX, RLi, RC≡CM, LiAlH4
-
Weaker nucleophiles
(neutral) require that the C≡N be activated prior to attack of the
Nu.
This can be done using a acid catalyst which protonates on the Lewis
basic N and makes the system more electrophilic.
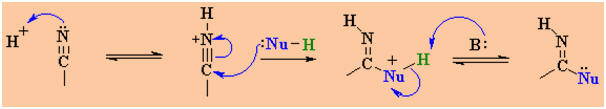
Examples of such nucleophilic systems are : H2O, ROH
|
The
protonation of a nitrile gives a structure that can be redrawn in
another resonance form that reveals the electrophilic character of
the C since it is a carbocation. |
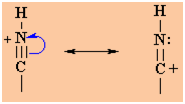
|
Back
to the top
Friedel-Crafts
Acylation of Benzene
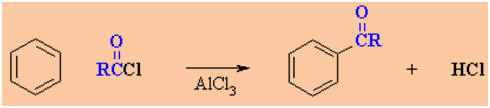
Reaction type: Electrophilic
Aromatic Substitution
Summary
-
Overall transformation : Ar-H
to Ar-COR(a ketone)
-
Named after Friedel and Crafts
who discovered the reaction.
-
Reagent : normally the acyl
halide (e.g. usually RCOCl) with aluminum trichloride, AlCl3,
a Lewis acid catalyst
-
The AlCl3 enhances
the electrophilicity of the acyl halide by complexing with the halide
-
Electrophilic species : the acyl
cation or acylium ion (i.e. RCO + ) formed by the
"removal" of the halide by the Lewis acid catalyst
-
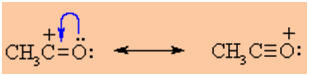
The acylium ion is stabilised by
resonance as shown below. This extra stability prevents the problems
associated with the rearrangement of simple carbocations:
-
The reduction of acylation
products can be used to give the equivalent of alkylation but avoids
the problems of rearrangement (more
details)
-
Friedel-Crafts reactions are
limited to arenes as or more reactive than mono-halobenzenes
-
Other sources of acylium can
also be used such as acid anhydrides with AlCl3
|
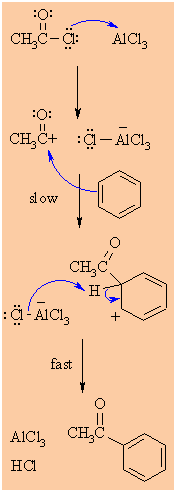
MECHANISM FOR THE FRIEDEL-CRAFTS ACYLATION OF BENZENE |
|
Step 1:
The acyl halide reacts with the Lewis acid to form a a more
electrophilic C, an acylium ion |
 |
|
Step 2:
The p electrons of the aromatic C=C act as a nucleophile,
attacking the electrophilic C+. This step destroys the aromaticity
giving the cyclohexadienyl cation intermediate. |
|
Step 3:
Removal of the proton from the sp3 C bearing the acyl-
group reforms the C=C and the aromatic system, generating HCl
and regenerating the active catalyst. |
Back
to the top
Hydrolysis of
Esters
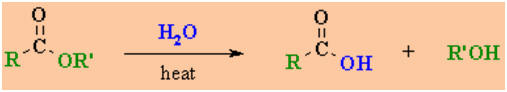
Reaction type: Nucleophilic
Acyl Substitution
Summary
-
Carboxylic esters hydrolyse to
the parent carboxylic acid and an alcohol.
-
Reagents : aqueous acid (e.g.
H2SO4) / heat,or aqueous NaOH / heat (known as "saponification").
-
These mechanisms are among some
of the most studied in organic chemistry.
-
Both are based on the formation
of a tetrahedral intermediate which then dissociates.
-
In both cases it is the C-O
bond between the acyl group and the oxygen that is cleaved.
Reaction under BASIC conditions:
-
The mechanism shown below leads
to acyl-oxygen cleavage (see step2).
-
The mechanism is supported by
experiments using 18O labeled compounds and esters of chiral
alcohols.
-
This reaction is known as "saponification"
because it is the basis of making soap from glycerol triesters in fats.
-
The mechanism is an example of
the reactive system type.
|
MECHANISM OF THE BASE
HYDROLYSIS OF ESTERS |
|
Step 1:
The hydroxide nucleophiles attacks at the electrophilic C
ofthe ester C=O, breaking the
p
bond and creating the tetrahedral intermediate. |
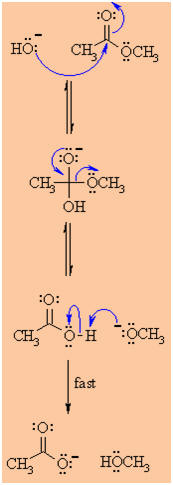
|
|
Step 2:
The intermediate collapses, reforming the C=O
results in the loss of the leaving group the alkoxide, RO-,
leading to the carboxylic acid. |
|
Step 3:
An acid / base reaction. A very rapid equilibrium where the
alkoxide,RO- functions as a base deprotonating the
carboxylic acid, RCO2H, (an acidic work up would
allow the carboxylic acid to be obtained from the reaction). |
| |
Reaction
under ACIDIC conditions:
-
Note that the acid catalysed
mechanism is the reverse of the Fischer esterification.
-
The mechanism shown below also
leads to acyl-oxygen cleavage (see step 5).
-
The mechanism is an example of
the less reactive system type.
|
MECHANISM OF THE ACID
CATALYSED HYDROLYSIS OF ESTERS |
|
Step 1:
An acid/base reaction. Since we only have a weak nucleophile and a
poor electrophile we need to activate the ester. Protonation of the
ester carbonyl makes it more electrophilic. |
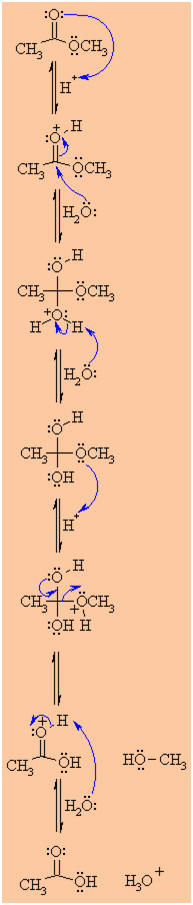
|
|
Step 2:
The water O functions as the nucleophile attacking the
electrophilic C in the C=O, with the electrons moving
towards the oxonium ion, creating the tetrahedral intermediate. |
|
Step 3:
An acid/base reaction. Deprotonate the oxygen that came from the
water molecule to neutralise the charge. |
|
Step 4:
An acid/base reaction. Need to make the -OCH3
leave, but need to convert it into a good leaving group first by
protonation. |
|
Step 5:
Use the electrons of an adjacent oxygen to help "push out" the
leaving group, a neutral methanol molecule. |
|
Step 6:
An acid/base reaction. Deprotonation of the oxonium ion reveals the
carbonyl C=O in the carboxylic acid product and regenerates
the acid catalyst. |
| |
Back
to the top
Preparation of Esters
Reaction
type: Nucleophilic Acyl Substiution
Summary
-
This reaction is also known as
the Fischer esterification.
-
Esters are obtained by refluxing
the parent carboxylic acid with the appropraite alcohol with an acid
catalyst.
-
The equilibrium can be driven to
completion by using an excess of either the alcohol or the carboxylic acid,
or by removing the water as it forms.
-
Alcohol reactivity order : CH3OH
> 1o > 2o > 3o (steric effects)
-
Esters can also be made from
other carboxylic acid derivatives, especially acyl halides and anhydrides,
by reacting them with the appropriate alcohol in the presence of a weak base
.
-
If a compound contains both
hydroxy- and carboxylic acid groups, then cyclic esters or lactones
can form via an intramolecular reaction. Reactions that form 5- or
6-membered rings are particularly favourable.
|
Study Tip:
The carboxylic acid and alcohol combination used to prepare an ester
are reflected by the name of the ester, e.g. ethyl acetate
(or ethyl ethanoate), CH3CO2CH2CH3
can be made from CH3CO2H, acetic acid (or
ethanoic acid) and HOCH2CH3 (ethanol). This
general "disconnection" is shown below:
 |
|
MECHANISM FOR REACTION
FOR ACID CATALYSED ESTERIFICATION
|
|
Step 1:
An acid/base reaction. Protonation of the carbonyl makes it more
electrophilic. |
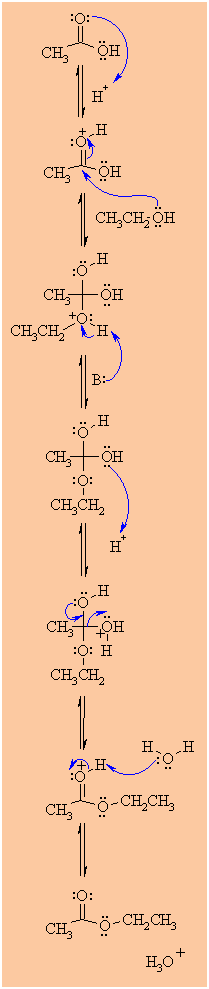 |
|
Step 2:
The alcohol O functions as the nucleophile attacking the
electrophilic C in the C=O, with the electrons moving
towards the oxonium ion, creating the tetrahedral intermediate. |
|
Step 3:
An acid/base reaction. Deprotonate the alcoholic oxygen. |
|
Step 4:
An acid/base reaction. Need to make an -OH leave, it doesn't
matter which one, so convert it into a good leaving group by
protonation. |
|
Step 5:
Use the electrons of an adjacent oxygen to help "push out" the
leaving group, a neutral water molecule. |
|
Step 6:
An acid/base reaction. Deprotonation of the oxonium ion reveals the
carbonyl in the ester product. |
Back
to the top
Reduction of
Esters
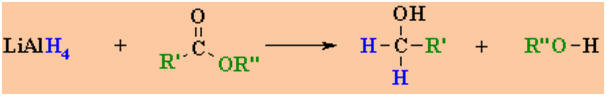
Reactions usually in Et2O or THF followed by H3O+work-ups
Reaction
type: Nucleophilic Acyl Substitution
then NucleophilicAddition
Summary
-
Carboxylic esters are reduced
give 2 alcohols, one from the alcohol portion of the ester and a 1o
alcohol from the reduction of the carboxylate portion.
-
Esters are less reactive towards
Nu than aldehydes or ketones.
-
They can only be reduced by
LiAlH4 but NOT by the less reactive NaBH4
-
The reaction requires that 2
hydrides (H-) be added to the carbonyl group of the ester
-
The mechanism is an example of
the reactive system type.
-
The reaction proceeds via a
aldehyde intermediate which then reacts with the second equivalent of the
hydride reagent (review)
-
Since the aldehyde is more
reactive than the ester, the reaction is not normally used as a preparation
of aldehydes .
|
MECHANISM OF THE
REACTION OF LiAlH4 WITH AN ESTER
|
|
Step 1:
The nucleophilic H from
the hydride reagent adds to the electrophilic C in the polar
carbonyl group of the ester. Electrons from the C=O move to
the electronegative O creating the tetrahedral
intermediate a metal alkoxide complex. |
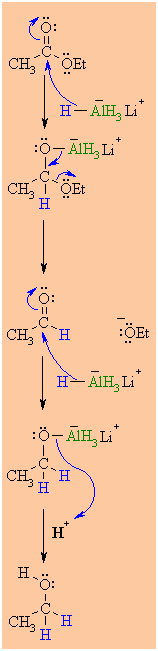 |
|
Step 2:
The tetrahedral intermediate collapses and displaces the alcohol
portion of the ester as a leaving group, in the form of the alkoxide,
RO-. This produces an aldehyde as an
intermediate. |
|
Step 3:
Now we are reducing an aldehyde (which we
have already seen)
The nucleophilic H from the hydride reagent adds to the
electrophilic C in the polar carbonyl group of the aldehyde.
Electrons from the C=O move to the electronegative O
creating an intermediate metal alkoxide complex. |
|
Step 4:
This is the work-up step, a simple acid/base reaction. Protonation
of the alkoxide oxygen creates the primary alcohol product from the
intermediate complex.
|
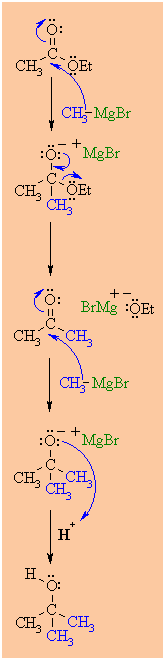
Reactions
of RLi and RMgX with Esters
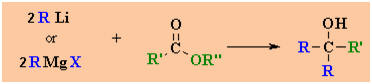
Reaction usually in Et2O followed by H3O+
work-up
Reaction type: Nucleophilic Acyl Substitution
then NucleophilicAddition
Summary
·
Carboxylic esters,
R'CO2R'', react with 2 equivalents of organolithium or Grignard
reagents to give tertiary alcohols.
·
The tertiary alcohol
that results contains 2 identical alkyl groups (from
R in the scheme)
·
The reaction proceeds
via a ketone intermediate which then reacts with the second equivalent of the
organometallic reagent (review)
·
Since the ketone is
more reactive than the ester, the reaction cannot be used as a
preparation of ketones.
·
The mechanism is an
example of the reactive system type.
|
MECHANISM OF THE REACTION OF RMgX WITH AN ESTER
|
|
Step 1:
The nucleophilic C in the organometallic reagent adds to
theelectrophilic C in the polar carbonyl group of the ester.
Electrons from the C=O move to the electronegative O
creating thetetrahderal intermediate, a metal alkoxide
complex. |
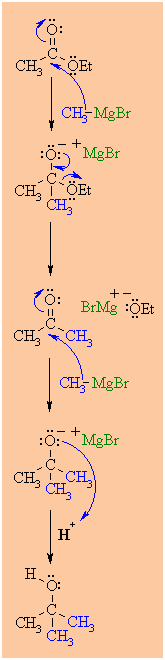
|
|
Step 2:
The tetrahedral intermediate collapses and displaces the alcohol
portion of the ester as a leaving group, in the form of the alkoxide,
RO-.This produces a ketone as an intermediate. |
|
Step 3:
The nucleophilic C in the organometallic reagent adds to the
electrophilic C in the polar carbonyl group of the ketone.
Electrons from the C=O move to the electronegative O
creating an intermediate metal alkoxide complex. |
|
Step 4:
This is the work-up step, a simple acid/base reaction.
Protonationof the alkoxide oxygen creates the alcohol product from
the intermediate complex.
|
Hydrolysis of
Amides
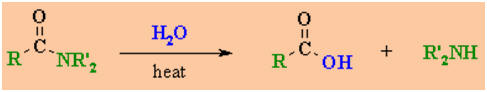
Reaction
type: Nucleophilic Acyl Substitution
Summary
-
Amides hydrolyse to the parent
carboxylic acid and the appropriate amine.
-
The mechanisms are similar to
those of esters.
-
Reagents : Strong acid (e.g.
H2SO4) / heat (preferred) or strong base (e.g.
NaOH) / heat.
Reaction under ACIDIC conditions:
-
Note that the acid catalysed
mechanism is analogous to the acid catalysed hydrolysis of esters.
-
The mechanism shown below
proceeds via protonation of the carbonyl not the amide N (see step 1).
-
The mechanism is an example of
the less reactive system type.
|
MECHANISM OF THE ACID
CATALYSED HYDROLYSIS OF AMIDES
|
|
Step 1:
An acid/base reaction. Since we only have a weak nucleophile and
apoor electrophile we need to activate the amide. Protonation of the
amidecarbonyl makes it more electrophilic. |
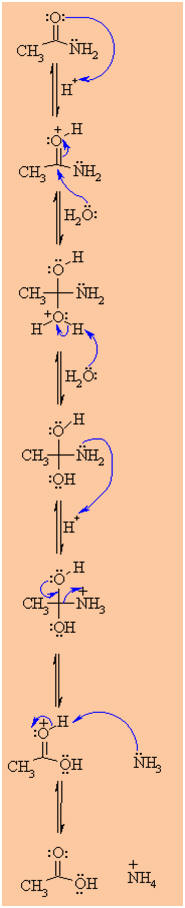
|
|
Step 2:
The water O functions as the nucleophile attacking the
electrophilicCin the C=O, with the electrons moving
towards the oxonium ion, creatingthe tetrahedral intermediate. |
|
Step 3:
An acid/base reaction. Deprotonate the oxygen that came from the
watermolecule to neutralise the charge. |
|
Step 4:
An acid/base reaction. Need to make the -NH2leave,
but need to convert it into a good leaving group first byprotonation. |
|
Step 5:
Use the electrons of an adjacent oxygen to help "push out" the
leavinggroup, a neutral ammonia molecule. |
|
Step 6:
An acid/base reaction. Deprotonation of the oxonium ion reveals
thecarbonyl in the carboxylic acid product and regenerates the acid
catalyst. |
| |
Back
to the top
Reduction of
Amides
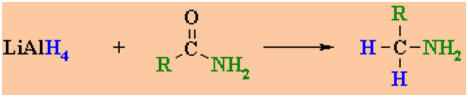
Reactions usually in Et2O or THF followed by H3O+
work-ups
Reaction type: Nucleophilic
Acyl Substitution then
Nucleophilic Addition
Summary
-
Amides, RCONR'2,
can be reduced to the amine, RCH2NR'2 by
conversion of the C=O to -CH2-
-
Amides can be reduced by LiAlH4
but NOT the less reactive NaBH4
-
Typical reagents : LiAlH4
/ ether solvent followedby aqueous work-up.
-
Note that this reaction is
different to that of other C=Ocompounds which reduce to
alcohols
-
The nature of the amine obtained
depends on the substituents present onthe original amide.
ook at the N substituents in the following examples (those bonds don'tchange
!)
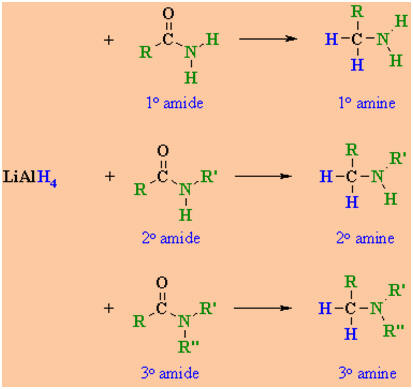
-
R,
R' or R" may be either alkyl or aryl substituents.
-
In the potential mechanism note
that it is an O system that leaves.This is consistent with O
systems being better leaving groups thatthe less electronegative N
systems.
|
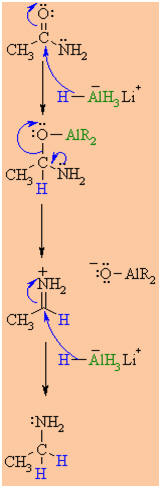
MECHANISM OF THE
REACTION OF LiAlH4 WITH AN AMIDE
|
|
Step 1:
The nucleophilic H from
the hydride reagent adds to the electrophilic C in the polar
carbonyl group of the ester. Electrons from the C=O move to
the electronegative O creating the tetrahedral
intermediate, a metal alkoxide complex. |
|
|
Step 2:
The tetrahedral intermediate collapses and displaces the O as
part of a metal alkoxide leaving group, this produces a highly
reactive iminium ion an intermediate. |
|
Step 3:
Rapid reduction by the nucleophilic
H from the hydride reagent as it adds to the
electrophilic C in the iminium system.
p
electrons from the
C=N move to the cationic N neutralising the charge
creating the amine product. |
|
|
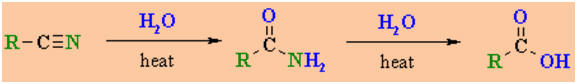
Hydrolysis of
Nitriles
Reaction
type: Nucleophilic Addition
then NucleophilicAcyl
Substitution
Summary
-
Nitriles, RC≡N, can be
hydrolysedto carboxylic acids, RCO2H via the amide,
RCONH2.
-
Reagents : Strong acid (e.g.
H2SO4) or strongbase (e.g. NaOH) / heat.
|
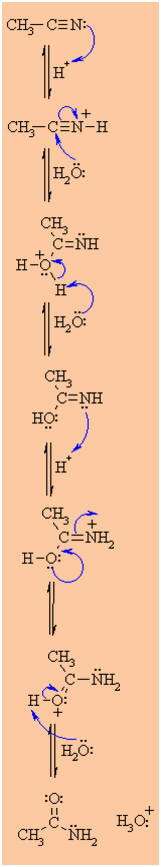
MECHANISM OF THE ACID CATALYSED HYDROLYSIS OF NITRILES
|
|
Step 1:
An acid/base reaction. Since we only have a weak nucleophile so
activate the nitrile, protonation makes it more electrophilic. |
|
|
Step 2:
The water O functions as the nucleophile attacking the
electrophilic C in the C≡N, with the electrons moving
towards the positive center. |
| |
|
Step 3:
An acid/base reaction. Deprotonate the oxygen that came from the
watermolecule. The remaining task is a
tautomerisation
at N and O centers. |
| |
|
Step 4:
An acid/base reaction. Protonate the N gives us the -NH2
we need.... |
|
Step 5:
Use the electrons of an adjacent O to neutralise the positive
at the N and form the
p
bond in the C=O. |
| |
|
Step 6:
An acid/base reaction. Deprotonation of the oxonium ion reveals the
carbonyl in the amide intermediate....halfway to the acid.....
|
| |
Reduction of
Nitriles
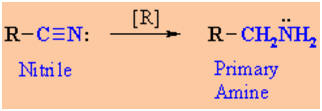
Reactions usually in Et2O or THF followed by H3O+work-up
Reaction type: Nucleophilic
Addition
Summary
-
The nitrile, RC≡N,
gives the 1o amine by conversion of the C≡N to -CH2-NH2
-
Nitriles can be reduced by LiAlH4
but NOT the less reactive NaBH4
-
Typical reagents : LiAlH4
/ ether solvent followed by aqueous work-up.
-
Catalytic hydrogenation (H2
/ catalyst) can also be used giving the same products.
-
R
may be either alkyl or aryl substituents
Reactions of RLi
or RMgX with Nitriles
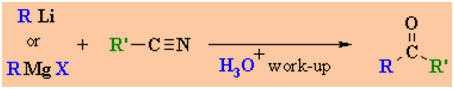
Reaction usually in Et2O or THF
Reaction type: Nucleophilic
Acyl Substitution then
Nucleophilic Addition
Summary:
-
Nitriles, RC≡N, react
with Grignard reagents or organolithium reagents to give ketones.
-
The strongly nucleophilic
organometallic reagents add to the C≡Nbond in a similar fashion to
that seen for
aldehydes and ketones.
-
The reaction proceeds via an
imine salt intermediate that is then hydrolysed to give the ketone product.
-
Since the ketone is not formed
until after the addition ofwater, the organometallic reagent
does not get the opportunity to
react with the ketone product.
-
Nitriles are less
reactive than aldehydes and ketones.
-
The mechanism is an example of
the reactive system type.
|
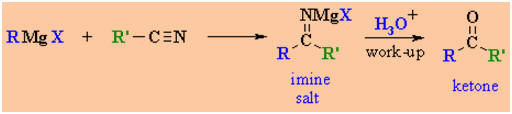
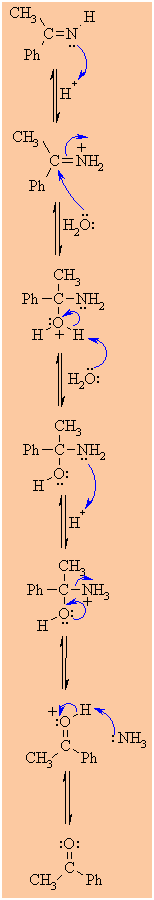
MECHANISM FOR THE REACTION OF RMgX WITH A NITRILE
|
|
Step 1:
The nucleophilic C in the organometallic reagent adds to
theelectrophilic C in the polar nitrile group. Electrons from
the C≡N move to the electronegative N creating an
intermediate imine salt complex. |
 |
|
Step 2:
An acid/base reaction. On addition of aqueous acid, the intermediate
salt protonates giving the imine. |
|
Step 3:
An acid/base reaction. Imines undergo nucleophilic addition,
but require activation by protonation (i.e. acid catalysis). |
|
Step 4:
Now the nucleophilic O of a water molecule attacks the
electrophilicCwith the
p
bond breaking to
neutralise the change on the N. |
| |
|
Step 5:
An acid/base reaction. Deprotonate the O from the water
molecule to neutralise the positive charge. |
|
Step 6:
An acid/base reaction. Before the N system leaves, it needs
to be made into a better leaving group by protonation. |
|
Step 7:
Use the electrons on the O in order to push out the N
leaving group, a neutral molecule of ammonia. |
| |
|
Step 8:
An acid/base reaction. Deprotonation reveals the carbonyl group
ofthe ketone product. |
Back
to the top
Spectroscopic Analysis
Spectroscopic
Analysis of Acyl Chlorides
-
IR
- presence of high
frequency C=O, C-Cl too low to be useful
|
Absorbance (cm-1)
|
Interpretation
|
|
1800 |
C=O stretch |
-
1H
NMR - only the
protons adjacent to the C=O are particularly characteristic.
|
Resonance (ppm)
|
Interpretation
|
|
~2 - 2.5 |
H-C-C=O |
-
13C
NMR
C=O typically 160-180 ppm (deshielding due to O)
-
minimal intensity,
characteristic of C's with no attached H's
-
UV-VIS
two absorption maxima p→p* (<200 nm) n→p* (~235 nm)
-
p electron from p of
C=O
-
n electron from O
lone pair
-
p* antibonding
C=O
-
Mass Spectrometry
Prominent peak corresponds to formation of acyl cations (acylium ions)

Spectroscopic
Analysis of Anhydrides
-
IR -
presence of two, high
frequency C=O
|
Absorbance (cm-1)
|
Interpretation
|
|
1820 |
C=O stretch |
|
1750 |
C=O stretch |
-
1H
NMR - only the
protons adjacent to the C=O are particularly characteristic.
|
Resonance (ppm)
|
Interpretation
|
|
~2 - 2.5 |
H-CC=O
|
-
13C
NMR
C=O typically 160-180 ppm (deshielding due to O)
-
minimal intensity,
characteristic of C's with no attached H's
-
UV-VIS
two absorption maxima p→p* (<200 nm) n→p* (~225nm,
diagnostic)
-
p electron from p of
C=O
-
n electron from O lone pair
-
p*
antibonding C=O
-
Mass Spectrometry
Prominent peak corresponds to formation of acyl cations (acylium ions)
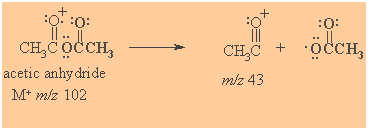
Spectroscopic
Analysis of Esters
-
IR
- presence of C=O,
and two C-O bands (Csp2-O and Csp3-O bonds)
|
Absorbance (cm-1)
|
Interpretation
|
|
1735 |
C=O stretch |
|
1300-1000 |
two bands for C-O stretch
|
-
1H
NMR -
deshielded proton of H-C-O is often recognisable, and H-C-C=O.
|
Resonance (ppm)
|
Interpretation
|
|
3.5-4.5 |
H-COC |
|
2-2.5 |
H-C-C=O |
-
13C
NMR
C=O typically 160-180 ppm (deshielding due to O)
-
minimal intensity,
characteristic of C's with no attached H's
-
UV-VIS
two absorption maxima p→p* (<200 nm) n→p* (~207 nm)
-
p electron from p of
C=O
-
n electron from O
lone pair
-
p*
antibonding C=O
-
Mass Spectrometry
Prominent peak corresponds to formation of acyl cations (acylium ions)
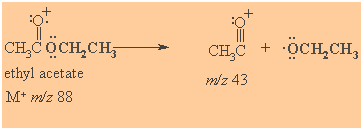
Spectroscopic
Analysis of Amides
-
IR
- presence of low
frequency C=O, N-H stretches for 1o or 2o amides.
|
Absorbance (cm-1)
|
Interpretation
|
|
1660 |
C=O stretch |
|
3500 and 3100 |
N-H stretch (two for NH2,
one for NH) |
-
1H
NMR - N-H
protons often broad,
|
Resonance (ppm)
|
Interpretation
|
|
5-8 (broad, exchangeable)
|
NH |
|
~ 2 - 2.4 |
H-C-C=O |
-
13C
NMR
C=O typically 160-180 ppm (deshielding due to O)
-
minimal intensity,
characteristic of C's with no attached H's
-
UV-VIS
absorption maxima n→p* (~215 nm)
-
n electron from O
lone pair
-
p*
antibonding C=O
-
Mass Spectrometry
Molecular ion M+ often visible.
A prominent peak corresponds to formation of acyl cations (acylium ions)
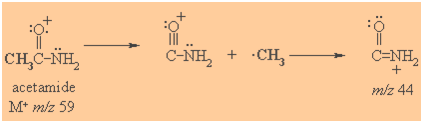
Spectroscopic
Analysis of Nitriles
-
IR
- very characteristic
C≡N stretch (only C≡C is similar region)
|
Absorbance (cm-1)
|
Interpretation
|
|
2250 |
C≡N stretch |
-
1H
NMR - only
protons adjacent to C≡N are likely to be characterisitic.
|
Resonance (ppm)
|
Interpretation
|
|
>
2 - 3 |
H-C-C≡N
|
-
13C
NMR
C≡N typically 115 -125 ppm (deshielding due to N)
-
minimal intensity,
characteristic of C's with no attached H's
-
UV-VIS
Simple nitriles usually show no absorption above 200 nm.
-
Mass Spectrometry
Molecular ion M+ is often weak or
absent, but a weak M-1 peak due to loss of an a-H is often present.
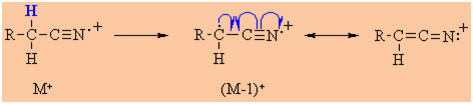
Back
to the top
 Alcohols
Alcohols
Aldehydes & Ketones
Alkyl Halide
Reaction
|