|
Introduction
An alkyl
halide is another name for a halogen-substituted
alkane. The carbon atom, which is bonded to the halogen
atom, has sp3 hybridized bonding orbitals and
exhibits a tetrahedral shape. Due to electronegativity
differences between the carbon and halogen atoms, the σ
covalent bond between these atoms is polarized, with the
carbon atom becoming slightly positive and the halogen atom
partially negative. Halogen atoms increase in size and
decrease in electronegativity going down the family in the
periodic table. Therefore, the bond length between carbon
and halogen becomes longer and less polar as the halogen
atom changes from fluorine to iodine.
POLARITY AND STRENGTH OF THE CARBON-X BONDS
-
Carbon-halogen bonds are very substantially polar
covalent, with carbon as the positive and halogen as the
negative end of the dipole. Conosequently, the carbon
attached to the halogen is electrophilic. We
shall see in the next chapter how nucleophiles react at
the carbon of an alkyl halide.
-
The carbon-fluorine bond is the strongest, especially
since fluorine is the most electronegative of the
halogens, resulting in a larger contribution of the
polar (ionic) structure to the resonance hybrid. The
larger contribution of the ionic structure not only
makes the molecule more polar, it also makes the bond
more stable because the ionic structure is lowered in
energy. However, all of the C-X bonds are significantly
polar.
-
The C-X bond dissociation energies (D), which you do not
need to memorize, are C-F 108; C-Cl 85; C-Br 70; C-I 57
(these are for CH3-X bonds).
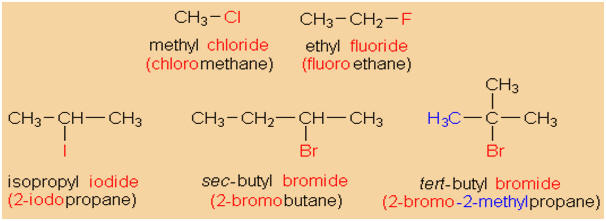
NOMENCLATURE
Alkyl halides are named using the
IUPAC rules for alkanes. Naming the alkyl group attached to
the halogen and adding the inorganic halide name for the
halogen atom creates common names.
-
Essentially, the naming of alkyl halides is not
different from the naming of alkanes. The halogen
atoms are treated as substituents on the main chain,
just as an alkyl group, and have no special priority
over alkyl groups.
-
The name of a chlorine substituent is "chloro", that
of a bromine substituent "bromo" and so on.
-
You sould practice naming a variety of haloalkanes.
Alkyl halides may formally be derived from alkanes
by exchanging hydrogen for a halogen atom (fluorine,
chlorine, bromine, or iodine). Alkyl halides are
classified into primary, secondary, and tertiary
alkyl halides, according to the degree of
substitution of the particular carbon atom that
carries the halogen.
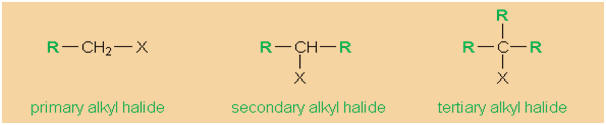
|
Primary, secondary, and tertiary alkyl
halides (X = F, Cl, Br, and I,
respectively). |
Vinyl halides are often classified as the fourth
type of alkyl halides.
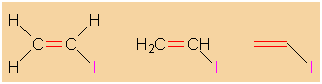
|
Different degrees of abstraction in the
illustration of vinyl iodide's
structure. |
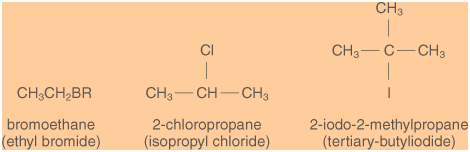
In the most generally accepted nomenclature of alkyl
halides the name of the alkyl residue is followed by
the halide's name, such as is the case with "methyl
iodide" and "ethyl chloride". In the IUPAC
nomenclature of alkyl halides (depicted in brackets
in the illustration below), an alkyl halide is
considered to be a substituted alkane. That is, the
name of the halogen is followed by the alkane's
name, such as, for example, "iodomethane" and
"chloromethane". If an alkyl halide contains more
than one halogen, the halogen names are noted in
alphabetical order, such as in
"1-chloro-2-iodobutane".
Examples of alkyl halide nomenclature.
Haloalkane style:
-
The root name is based on the longest chain containing
the halogen.
-
This root give the alkane part of the name.
-
The type of halogen defines the halo prefix, e.g.
chloro-
-
The chain is numbered so as to give the halogen the
lowest possible number
Alkyl halide style:
-
The root name is based on the longest chain containing
the halogen.
-
This root give the alkyl part of the name.
-
The type of halogen defines the halide suffix, e.g.
chloride
-
The chain is numbered so as to give the halogen the
lowest possible number.
|
Haloalkane style:
-
Functional group is an alkane, therefore
suffix = -ane
-
The longest continuous chain is C3 therefore
root = prop
-
The substituent is a chlorine, therefore
prefix = chloro
-
The first point of difference rule requires
numbering from the right as drawn,
the substituent locant is 1-
1-chloropropane
|

CH3CH2CH2Cl |
|
Alkyl halide style:
-
The alkyl group is C4, it's a tert-butyl
-
The halogen is a bromine, therefore suffix =
bromide
tert-butyl
bromide
Haloalkane style:
-
Functional group is an alkane, therefore
suffix = -ane
-
The longest continuous chain is C3 therefore
root = prop
-
The substituent is a bromine, therefore
prefix = bromo
-
There is a C1 substituent = methyl
-
The substituent locants are both 2-
2-bromo-2-methylpropane |
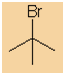
(CH3)3CBr |
|
Haloalkane style:
-
Functional group is an alkene, therefore
suffix = -ene
-
The longest continuous chain is C4 therefore
root = but
-
The substituent is a bromine, therefore
prefix = bromo
-
Since bromine is named as a substituent, the
alkene gets priority
-
The first point of difference rule requires
numbering from the left as drawn to
make the alkene group locant 1-
-
Therefore the bromine locant 4-
4-bromobut-1-ene |
CH2=CHCH2CH2Br |
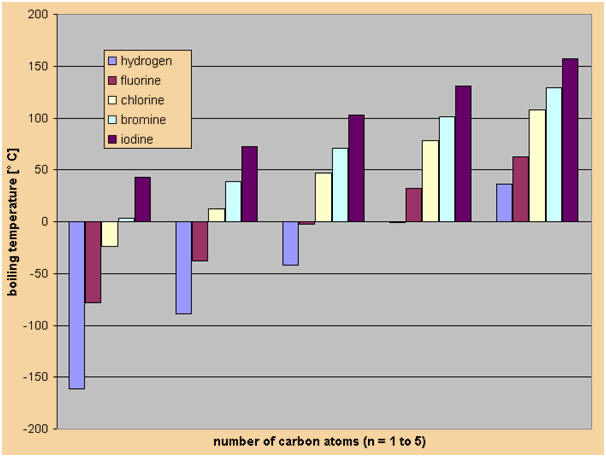
Physical properties
The physical properties of alkyl halides differ considerably
from that of the corresponding alkanes. The strength and
length of the carbon-halogen bond and the dipole moments and
boling points of alkyl halides are determined by the bond's
polarity, as well as the size of the various halogen atoms:
�
The C-X bond strength decreases with an increase in the size
of the halogen (X), because the size of the halogen's p
orbital increases, as well. Thus, the p orbital becomes
hazier, and the overlap with the carbon's orbital
deteriorates. As a result, the C-X bond is weakend and
elongated.
Bond lenghts,
dipole moments, and dissociation energies of methyl halidies
( CH3X)
|
Methyl halide (halomethan) |
|
|
|
|
|
Bond length (pm) |
158.5 |
178.4 |
192.9 |
213.9 |
|
Dipole moment (D) |
1.85 |
1.87 |
1.81 |
1.62 |
|
Dissociation energy (kj/mol) |
416 |
356 |
297 |
239 |
�
Halogens (F, Cl, and Br) are comparably more electronegative
than carbon is. Consequently, carbon atoms that carry
halogens are partially positively charged while the halogen
is partially negatively charged. The polarity of the C-X
bond causes a measureable dipole moment. As a result of the
partial positive charge, the carbon atom displays an
electrophilic character. The chemical behaviour of alkyl
halides is mainly determined by the carbon's
electrophilicity.
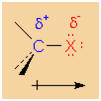
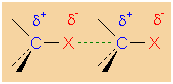
Polar character of a C-X bond
Dipole-dipole interaction in alky halides
�
The boiling points of alkyl halides are considerably
higher than that of the corresponding alkanes. The main
reason for this is the dipole moment of alkyl halides,
which leads to attractive dipole-dipole interactions in
liquid alkyl halides. Furthermore, the higher molar mass
and the stronger London forces (Cl, Br, and I) lead to
higher boiling points. The main reason of the stronger
London forces between alkyl halides is the fact that the
electron shell of halogens is larger than that of
hydrogen and carbon. In larger electron shells, the
electrons are not as strongly attracted by the nucleus
as in small electron shells. Consequently, the
interactions between the electron shells of larger atoms
are stronge
Boiling points of alkanes (X = H) and alkyl halides (X =
F, Cl, Br, I) (in �C).
Structure:
-
The alkyl halide functional group consists of an sp3
hybridised C atom bonded to a halogen, X, via a σ
bond.
-
The carbon halogen bonds are typically quite polar
due to the electronegativity and polarisability of
the halogen.
Reactivity:
-
The halogens (Cl, Br and I) are good leaving groups.
-
The polarity makes the C atom electrophilic and
prone to attack by nucleophiles via
SN1 or
SN2 reactions.
-
Bases can remove β-hydrogens and cause
1,2-elimination to form alkenes via E1 or E2
reactions.
-
Insertion of a metal (esp. Mg) creates an
organometalic species.
Preparation
Alkyl halides may be synthesized by
addition, as well as by
substitution reactions:
�
Addition of a hydrogen halide HX ( HX= HCl,
HBr or HI)
to an alkene yields the corresponding monohalogenated alkene
(Markovnikv addition). The addition of bromine and chlorine
to alkenes results in the corresponding vicinal alykl
dihalides.
|
Electrophilic
addition of hydrogen halides and halogens to
1-methylcyclopentene. |
|
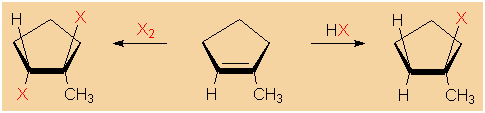 |
�
The
radical substitution of an alkane's hydrogen with
bromine or chlorine is yet another method of synthesizing
alkyl halides. However, the praticality of this method is
limited, as mixtures of alkyl halides with varying degree of
halogenation are obtained.
E HALOGENATION REACTION
-
A very simple example of the halogenation of alkanes is
the chlorination of methane, as shown in the
illustration below. The products are HCl and
chloromethane.
-
The REACTION TYPE is SUBSTITUTION, since a
hydrogen of methane is replaced by a chlorine atom. The
MECHANISTIC TYPE, as we will see, is Homolytic
or Radical. The overall designation of the reaction,
then, is SH (S WITH A SUBSCRIPT H).
-
Virtually any C-H bond in which the carbon atom is
tetrahedrally hybridized can be chlorinated, so that
chloromethane can be further converted to
dichloromethane, and this on to trichloromethane
(chloroform), and finally to tetrachloromethane (carbon
tetrachloride). You may recognize these chlorinated
compounds as common solvents, both in the laboratory and
in commercial uses.
-
In the chlorination of alkanes more complex than methane
or ethane, more than one monochloroalkane can be formed.
We will refer to the preference for the formation of one
constitutional isomer over the other as regiospecificity.
For example, propane can be converted to both
1-chloropropane and 2-chloropropane. Actually, both are
formed, but the 2-chloropropane is slightly, but only
slightly, preferred. Thus, the reaction is not very
stereooregiospecific.
|
Radical chlorination of methane. |
|
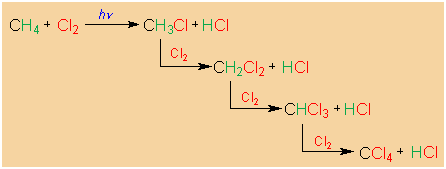 |
Mechanism
|
Simplifying all this for exam purposes:
Initiation Step:
|
1- |
Br2��>2Br |
Propagation Step:
|
2- |
CH4 + Br��>CH3
+ HBr |
|
3- |
CH3
+ Br2��>CH3Br
+ Br |
Termination Step:
|
4- 2Br��>Br2
|
|
5- |
CH3
+ Br��>CH3Br
|
|
6- |
CH3
+ CH3��>CH3CH3
|
|
|
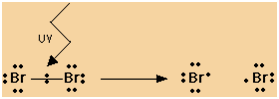 |
| |
|
| |
Br2��>2Br |
| |
Br
+ Br-Br��>Br-Br
+ Br
|
|
| |
|
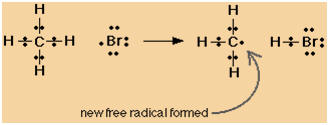
| |
CH4 + Br��>CH3
+ HBr |
.
| |
CH3
+ Br��>CH3Br
|
| |
CH3
+ CH3��>CH3CH3
|
| |
CH3
+ Br2��>CH3Br
+ Br |
|
|
-
This is our first example of a reaction mechanism in
which radicals are involved. The definition of a
radical is any species which has an unpaired or odd
electron. There are two radical species involved
in this mechanism, the chlorine atom and the methyl
radical. The naming of organic radicals is
simple, it is essentially the name of the
corresponding substituent with the name radical
being appended to it.
-
The overall reaction is said to be a homolytic
substitution reaction, as noted previously,
because the bonds which are broken are broken
homolytically, i.e., one electron departing with
each component of the bond. In homolytic cleavages
radicals are always formed, so the reaction
mechanism can also be called radical substitution.
-
One specific way in which radical reactions can
occur is by means of a radical chain reaction.
It is important to keep in mind that not all
radical reaction mechanisms are radical chain
mechanisms. A radical chain mechanism is one in
which a particular set or two or three steps is
repeated over and over without the necessity of
generating more radicals. This is seen in steps 2
and 3 of the mechanism above. The stage of the
reaction which represents the chain is called the
propagation cycle. It is so called because in
it, the observed products (HCl and chloromethane)
are propagated or made. An efficient set of
progpagation reactions is essential to a successful
radical chain mechanism.
-
Overall, this or any, radical chain mechanism
consists of three discrete stages or parts.
The first part is called initiation. In the
initiation stage of the mechanism (step 1), the
radicals which are necessary to enter the
propagation cycle are generated. This typically
involves the homolytic cleavage of a covalent bond,
and so it requires energy. In this case, it is the
Cl-Cl bond which is cleavaged homolytically.It is
for this reason that the chlorination reaction
requires the input of either heat or photochemical
energy.
-
As noted above, the second stage or part of the
radical chain mechanism is the propagation cycle.
An efficient propagation cycle uses a relatively
few radicals to generate a large amount of product.
It is desirable that for each radical produced in
the initiation reaction, hundreds or even thousands
of product molecules be generated before the radical
is destroyed.
-
The third and final stage of the radical chain
mechanism is termination. Termination is the
undesirable but unavoidable coupling between two
radicals which destroys the chain carrying radicals
and stops the current radical chain. It is thus the
competitor of propagation. Because of this
continuing consumption of radicals, initiation must
continue to progressively generate more radicals.
For a chain reaction to be effective, propagation by
the radicals must be much more efficient than
coupling between them.These coupling reactions are
extrememly fast, because no bond is broken and one
bond is formed. However, since the radical
concentrations are extremely small, the probability
of one radical meeting another is much less than for
a radical to meet a molecule of chlorine or methane.
�
Nevertheless, a selective monohalogenation in allylic
position may be achieved by applying N-bromosuccinimide (NBS).
This method was introduced by Karl Ziegler in 1942.
|
Bromination of cyclohexene in allylic position. |
|
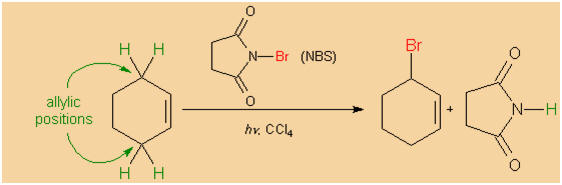 |
�
A standard method of synthesizing alkyl halides is the
treatment of alcohols with HCl, HBr or BI.
The reaction is a nucleophilic substitution in which the
alcohol's hydroxy group is exchanged for the halide ion.
Hydroxide is a poor leaving group though it may be converted
into the good leaving group water through protonation by a
hydrogen halide. However, at moderate temperature, the
reaction is practicable only with tertiary alcohols. A
higher reaction temperature is required if the reaction
ought to be carried out with primary or secondary alcohols.
Otherwise, the reaction rate will be too low. In contrast,
the reaction of tertiary alcohols with hydrogen halides is
much more rapid. As a result, considerable conversion is
obtained within a period of only a few minutes when pure
HCl, or HBr is passed through the alcohol. However, the
hydrogen halides in alkyl halides' syntheses are more
frequently generated in situ by treating the halide ion with
phosphoric or sulfuric acid.
|
Alkyl halide synthesis by treatment of alcohols
with HX. |
|
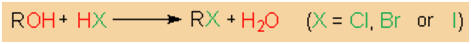
|
|
Chlorination of 1-methylcyclohexanol. |
|
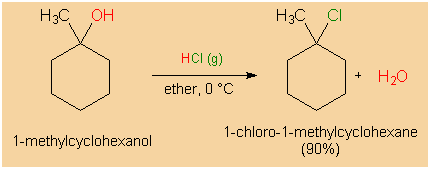 |
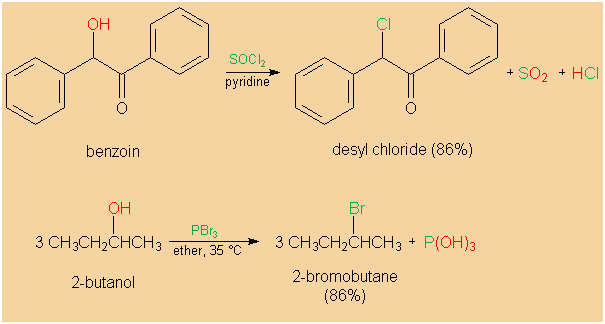
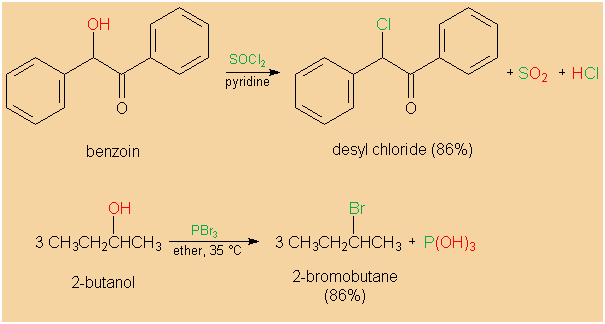
HBr and HI are usually used for the synthesis of alkyl
bromides and iodides, respectively. However, PBr3 may
also be applied. Aside from HCl, inorganic acid halides,
such as thionyl chloride (SOCl2),
phosphorus trichloride (PCl3),
phosphorus pentachloride (PCl5),
or phosphorus oxychloride (POCl3),
are common chlorinating agents in alkyl chlorides'
syntheses. If such chlorination agents are employed in the
conversion of alcohols into alkyl halides, rearrangements
are much less often the case than with HCl.
|
Halogenation of secondary alcohols by inorganic
acid halides. |
|
 |
|
|
Reaction of
Alkyl Halides
The functional group of alkyl halides is a carbon-halogen bond, the
common halogens being fluorine, chlorine, bromine and iodine. With the
exception of iodine, these halogens have electronegativities
significantly greater than carbon. Consequently, this functional group
is polarized so that the carbon is electrophilic and the halogen is
nucleophilic, as shown in the drawing on the right. Two characteristics
other than electronegativity also have an important influence on the
chemical behavior of these compounds. The first of these is covalent
bond strength. The strongest of the carbon-halogen covalent bonds is
that to fluorine. Remarkably, this is the strongest common single bond
to carbon, being roughly 30 kcal/mole stronger than a carbon-carbon bond
and about 15 kcal/mole stronger than a carbon-hydrogen bond. Because of
this, alkyl fluorides and fluorocarbons in general are chemically and
thermodynamically quite stable, and do not share any of the
reactivity patterns shown by the other alkyl halides. The
carbon-chlorine covalent bond is slightly weaker than a carbon-carbon
bond, and the bonds to the other halogens are weaker still, the bond to
iodine being about 33% weaker. The second factor to be considered is the
relative stability of the corresponding halide anions, which is
likely the form in which these electronegative atoms will be replaced.
This stability may be estimated from the relative acidities of the H-X
acids, assuming that the strongest acid releases the most stable
conjugate base (halide anion). With the exception of HF (pKa
= 3.2), all the hydrohalic acids are very strong, small differences
being in the direction HCl < HBr < HI.
Reactions at
sp�-hybridized
Carbons: Substitution and Elimination
Fundamentally, organic compounds that contain a carbon with a more
electronegative substituent display two types of reaction: nucleophilic
substitution reactions and elimination reactions. In nucleophilic
substitution, the electronegative substituent (leaving group) is
exchanged (substituted) for another substitutent (nucleophile). In
elimination however, the electronegative substituent is released from
the organic molecule along with another atom or group, which is usually
a hydrogen from the vicinal carbon.
Alkyl halides (R-X) are characteristic examples of such organic compounds. The
leaving groups in nucleophilic substitutions and eliminations with alkyl halides
are the halide anions ((Xˉ)
Cl ˉ,
Br ˉ,
I ˉand
F ˉ.
virtually never functions as a leaving group.
|
Reaction types of alkyl halides. |
|

|
Substitution reactions are particularly useful in organic chemistry - though
this is not only because they enable the synthetically easily available alkyl
halides to be converted into multiple other organic compounds.
Substitution and Elimination
The characteristics noted above lead us to anticipate certain types of
reactions that are likely to occur with alkyl halides. In describing
these, it is useful to designate the halogen-bearing carbon as alpha
and the carbon atom(s) adjacent to it as beta, as noted in the
first four equations shown below. Replacement or substitution of the
halogen on the α-carbon (colored maroon) by a nucleophilic reagent is a
commonly observed reaction, as shown in equations
1, 2, 5, 6 & 7
below. Also, since the electrophilic character introduced by the halogen
extends to the β-carbons, and since nucleophiles are also bases, the
possibility of base induced H-X elimination must also be considered, as
illustrated by equation
3.
Finally, there are some combinations of alkyl halides and nucleophiles
that fail to show any reaction over a 24 hour period, such as the
example in equation
4.
For consistency, alkyl bromides have been used in these examples.
Similar reactions occur when alkyl chlorides or iodides are used, but
the speed of the reactions and the exact distribution of products will
change.

In order to understand why some combinations of alkyl halides and
nucleophiles give a substitution reaction, whereas other combinations
give elimination, and still others give no observable reaction, we must
investigate systematically the way in which changes in reaction
variables perturb the course of the reaction. The following general
equation summarizes the factors that will be important in such an
investigation.

One conclusion, relating the structure of the R-group to possible
products, should be immediately obvious. If R- has no beta-hydrogens
an elimination reaction is not possible, unless a structural
rearrangement occurs first. The first four halides shown on the left
below do not give elimination reactions on treatment with base, because
they have no β-hydrogens. The two halides on the right do not normally
undergo such reactions because the potential elimination products have
highly strained double or triple bonds.
It is also worth noting that sp2
hybridized C�X compounds, such as the three on the right, do not
normally undergo nucleophilic substitution reactions, unless other
functional groups perturb the double bond(s).

Using the general reaction shown above as our reference, we can identify
the following variables and observables.
|
Variables |
R
change α-carbon from 1� to 2� to 3�
if the α-carbon is a chiral center, set as (R) or
(S)
X
change from Cl to Br to I (F is relatively unreactive)
Nu:
change from anion to neutral; change basicity; change
polarizability
Solvent
polar vs. non-polar; protic vs. non-protic |
|
Observables |
Products
substitution, elimination, no reaction.
Stereospecificity if the α-carbon is a chiral
center what happens to its configuration?
Reaction Rate measure as a function of reactant
concentration. |
When several reaction variables may be changed, it is important to
isolate the effects of each during the course of study. In other words:
only one variable should be changed at a time, the others being
held as constant as possible. For example, we can examine the effect of
changing the halogen substituent from Cl to Br to I, using ethyl as a
common R�group, cyanide anion as a common nucleophile, and ethanol as a
common solvent. We would find a common substitution product, C2H5�CN,
in all cases, but the speed or rate of the reaction would increase in
the order: Cl < Br < I. This reactivity order reflects both the strength
of the C�X bond, and the stability of X(�)
as a leaving group, and leads to the general conclusion that alkyl
iodides are the most reactive members of this functional class.
1. Nucleophilicity
Recall the definitions of electrophile and nucleophile:
Electrophile:
An electron deficient atom, ion or molecule that has an affinity for
an electron pair, and will bond to a base or nucleophile.
Nucleophile:
An atom, ion or molecule that has an electron pair that may be donated
in forming a covalent bond to an electrophile (or Lewis acid).
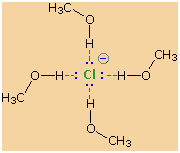
If we use a common alkyl halide, such as methyl bromide, and a common
solvent, ethanol, we can examine the rate at which various nucleophiles
substitute the methyl carbon. Nucleophilicity is thereby related
to the relative rate of substitution reactions at the halogen-bearing
carbon atom of the reference alkyl halide. The most reactive
nucleophiles are said to be more nucleophilic than less reactive members
of the group. The nucleophilicities of some common Nu:(�)
reactants vary as shown in the following chart.
Nucleophilicity:
CH3CO2(�)
< Cl(�)
< Br(�)
< N3(�)
< CH3O(�)
< CN(�)
< I(�)
< SCN(�)
< I(�)
< CH3S(�)

The reactivity range encompassed by these reagents is over 5,000 fold,
thiolate being the most reactive. Note that by using methyl bromide as
the reference substrate, the complication of competing elimination
reactions is avoided. The nucleophiles used in this study were all
anions, but this is not a necessary requirement for these substitution
reactions. Indeed reactions
6 & 7,
presented at the beginning of this section, are examples of neutral
nucleophiles participating in substitution reactions. The cumulative
results of studies of this kind has led to useful empirical rules
pertaining to nucleophilicity:
(i)
For a given element, negatively charged species are more nucleophilic
(and basic) than are equivalent neutral species.
(ii) For a given period of the periodic table, nucleophilicity
(and basicity) decreases on moving from left to right.
(iii) For a given group of the periodic table, nucleophilicity
increases from top to bottom (i.e. with increasing size),
although there is a solvent dependence due to hydrogen bonding. Basicity
varies in the opposite manner.
2. Solvent Effects
Solvation
of nucleophilic anions markedly influences their reactivity. The
nucleophilicities cited above were obtained from reactions in methanol
solution. Polar, protic solvents such as water and alcohols solvate
anions by hydrogen bonding interactions, as shown in the diagram on the
right. These solvated species are more stable and less reactive than the
unsolvated "naked" anions. Polar, aprotic solvents such as DMSO
(dimethyl sulfoxide), DMF (dimethylformamide) and acetonitrile do not
solvate anions nearly as well as methanol, but provide good solvation of
the accompanying cations. Consequently, most of the nucleophiles
discussed here react more rapidly in solutions prepared from these
solvents. These solvent effects are more pronounced for small basic
anions than for large weakly basic anions. Thus, for reaction in DMSO
solution we observe the following reactivity order:
Nucleophilicity:
I(�)
< SCN(�)
< Br(�)
< Cl(�)
≈ N3(�)
< CH3CO2
(�)
< CN(�)
≈ CH3S(�)
< CH3O(�)

Note that this order is roughly the order of increasing basicity.
3. The Alkyl Moiety
Some of the most important information concerning nucleophilic
substitution and elimination reactions of alkyl halides has come from
studies in which the structure of the alkyl group has been varied. If we
examine a series of alkyl bromide substitution reactions with the strong
nucleophile thiocyanide (SCN) in ethanol solvent, we find large
decreases in the rates of reaction as alkyl substitution of the
alpha-carbon increases. Methyl bromide reacts 20 to 30 times faster than
simple 1�-alkyl bromides, which in turn react about 20 times faster than
simple 2�-alkyl bromides, and 3�-alkyl bromides are essentially
unreactive or undergo elimination reactions. Furthermore, β-alkyl
substitution also decreases the rate of substitution, as witnessed by
the failure of neopentyl bromide, (CH3)3CCH2-Br
(a 1�-bromide), to react.
Alkyl halides in which the alpha-carbon is a chiral center provide
additional information about these nucleophilic substitution reactions.
Returning to the examples presented at the beginning of this section, we
find that reactions
2, 5 & 6
demonstrate an inversion of configuration when the cyanide nucleophile
replaces the bromine. Other investigations have shown this to be
generally true for reactions carried out in non-polar organic solvents,
the reaction of (S)-2-iodobutane with sodium azide in ethanol being just
one example ( in the following equation the alpha-carbon is maroon and
the azide nucleophile is blue). Inversion of configuration during
nucleophilic substitution has also been confirmed for chiral 1�-halides
of the type RCDH-X,
where the chirality is due to isotopic substitution.
(S)-CH3CHICH2CH3
+ NaN3
��>
(R)-CH3CHN3CH2CH3
+ NaI
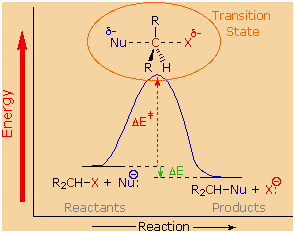
We can now piece together a plausible picture of how nucleophilic
substitution reactions of 1� and 2�-alkyl halides take place. The
nucleophile must approach the electrophilic alpha-carbon atom from the
side opposite the halogen. As a covalent bond begins to form between the
nucleophile and the carbon, the carbon halogen bond weakens and
stretches, the halogen atom eventually leaving as an anion. The diagram
on the right shows this process for an anionic nucleophile. We call this
description the SN2
mechanism, where S stands for Substitution, N
stands for Nucleophilic and 2 stands for bimolecular
(defined below). In the SN2
transition state the alpha-carbon is hybridized sp2 with
the partial bonds to the nucleophile and the halogen having largely
p-character. Both the nucleophile and the halogen bear a partial
negative charge, the full charge being transferred to the halogen in the
products. The consequence of rear-side bonding by the nucleophile is an
inversion of configuration about the alpha-carbon. Neutral nucleophiles
react by a similar mechanism, but the charge distribution in the
transition state is very different.
This mechanistic model explains many aspects of the reaction. First, it
accounts for the fact that different nucleophilic reagents react at very
different rates, even with the same alkyl halide. Since the transition
state has a partial bond from the alpha-carbon to the nucleophile,
variations in these bond strengths will clearly affect the activation
energy, ΔE�,
of the reaction and therefore its rate. Second, the rear-side approach
of the nucleophile to the alpha-carbon will be subject to hindrance by
neighboring alkyl substituents, both on the alpha and the beta-carbons.
The following models clearly show this "steric hindrance" effect.
The two models displayed below start as methyl bromide, on the left,
and ethyl bromide, on the right. These may be replaced by
isopropyl, tert-butyl, neopentyl, and benzyl bromide models by pressing
the appropriate buttons. (note that when first activated, this display
may require clicking twice on the selected button.) In each picture the
nucleophile is designated by a large deep violet sphere, located 3.75
Angstroms from the alpha-carbon atom (colored a dark gray), and located
exactly opposite to the bromine (colored red-brown). This represents a
point on the trajectory the nucleophile must follow if it is to bond to
the back-side of the carbon atom, displacing bromide anion from the
front face. With the exception of methyl and benzyl, the other alkyl
groups present a steric hindrance to the back-side approach of the
nucleophile, which increases with substitution alpha and beta to the
bromine. The hydrogen (and carbon) atoms that hinder the nucleophile's
approach are colored a light red. The magnitude of this steric hindrance
may be seen by moving the models about in the usual way, and is clearly
greatest for tert-butyl and neopentyl, the two compounds that fail to
give substitution reactions.
4. Molecularity
If a chemical reaction proceeds by more than one step or stage, its
overall velocity or rate is limited by the slowest step, the
rate-determining step. This "bottleneck concept" has analogies in
everyday life. For example, if a crowd is leaving a theater through a
single exit door, the time it takes to empty the building is a function
of the number of people who can move through the door per second. Once a
group gathers at the door, the speed at which other people leave their
seats and move along the aisles has no influence on the overall exit
rate. When we describe the mechanism of a chemical reaction, it is
important to identify the rate-determining step and to determine its "molecularity".
The molecularity of a reaction is defined as the number of
molecules or ions that participate in the rate determinining step. A
mechanism in which two reacting species combine in the transition state
of the rate-determining step is called bimolecular. If a single
species makes up the transition state, the reaction would be called
unimolecular. The relatively improbable case of three independent
species coming together in the transition state would be called
termolecular.
5. Kinetics
One way of investigating the molecularity of a given reaction is to
measure changes in the rate at which products are formed or reactants
are lost, as reactant concentrations are varied in a systematic fashion.
This sort of study is called
kinetics,
and the goal is to write an equation that correlates the observed
results. Such an equation is termed a kinetic expression, and for a
reaction of the type: A + B ���> C + D it takes the form: Reaction
Rate = k[A]
n[B]
m,
where the rate constant k is a proportionality constant that
reflects the nature of the reaction, [A] is the concentration of
reactant A, [B] is the concentration of reactant B, and n
& m are exponential numbers used to fit the rate equation to the
experimental data. Chemists refer to the sum n + m as the kinetic
order of a reaction. In a simple bimolecular reaction n & m would
both be 1, and the reaction would be termed second order,
supporting a mechanism in which a molecule of reactant A and one of B
are incorporated in the transition state of the rate-determining step. A
bimolecular reaction in which two molecules of reactant A (and no B) are
present in the transition state would be expected to give a kinetic
equation in which n=2 and m=0 (also second order). The kinetic
expressions found for the reactions shown at the beginning of this
section are written in blue in the following equations. Each different
reaction has its own distinct rate constant, k#.
All the reactions save
7
display second order kinetics, reaction
7
is first order.

It should be recognized and remembered that the molecularity of a
reaction is a theoretical term referring to a specific mechanism. On the
other hand, the kinetic order of a reaction is an experimentally derived
number. In ideal situations these two should be the same, and in most of
the above reactions this is so. Reaction
7
above is clearly different from the other cases reported here. It not
only shows first order kinetics (only the alkyl halide concentration
influences the rate), but the chiral 3�-alkyl bromide reactant undergoes
substitution by the modest nucleophile water with extensive racemization.
Note that the acetonitrile cosolvent does not function as a nucleophile.
It serves only to provide a homogeneous solution, since the alkyl halide
is relatively insoluble in pure water.
One of the challenges faced by early workers in this field was to
explain these and other differences in a rational manner.
Two discrete mechanisms for nucleophilic substitution reactions will be
described in the next section.
Reactions of Alkyl Halides with
Reducing Metals
The alkali metals (Li, Na, K etc.) and the alkaline earth metals
(Mg and Ca, together with Zn) are good reducing agents, the former being
stronger than the latter. Sodium, for example, reduces elemental
chlorine to chloride anion (sodium is oxidized to its cation), as do the
other metals under varying conditions. In a similar fashion these same
metals reduce the carbon-halogen bonds of alkyl halides. The halogen is
converted to halide anion, and the carbon bonds to the metal (the carbon
has carbanionic character). Halide reactivity increases in the order: Cl
< Br < I. The following equations illustrate these reactions for the
commonly used metals lithium and magnesium (R may be hydrogen or alkyl
groups in any combination). The alkyl magnesium halides described in the
second reaction are called Grignard Reagents after the French
chemist who discovered them. The other metals mentioned above react in a
similar manner, but the two shown here are the most widely used.
Although the formulas drawn here for the alkyl lithium and Grignard
reagents reflect the stoichiometry of the reactions and are widely used
in the chemical literature, they do not accurately depict the structural
nature of these remarkable substances. Mixtures of polymeric and other
associated and complexed species are in equilibrium under the conditions
normally used for their preparation.
|
R3C-X
+ 2Li
��>
R3C-Li +
LiX
An Alkyl Lithium Reagent
R3C-X +
Mg
��>
R3C-MgX A Grignard Reagent |
The metals referred to here are insoluble in most organic solvents,
hence these reactions are clearly heterogeneous, i.e. take place on the
metal surface. The conditions necessary to achieve a successful reaction
are critical.
First, the metal must be clean and finely divided so as to
provide the largest possible surface area for reaction.
Second, a suitable solvent must be used. For alkyl lithium
formation pentane, hexane or ethyl ether may be used; but ethyl ether or
THF are essential for Grignard reagent formation.
Third, since these organometallic compounds are very reactive,
contaminants such as water, alcohols and oxygen must be avoided.
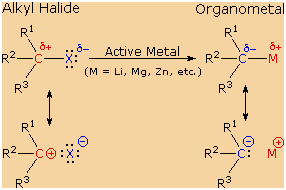
These reactions are obviously substitution reactions, but they
cannot be classified as nucleophilic substitutions, as were the earlier
reactions of alkyl halides. Because the functional carbon atom has been
reduced, the polarity of the resulting functional group is inverted (an
originally electrophilic carbon becomes nucleophilic). This change,
shown below, makes alkyl lithium and Grignard reagents unique and useful
reactants in synthesis.
Reactions of organolithium and Grignard reagents reflect the
nucleophilic (and basic) character of the functional carbon in these
compounds. Many examples of such reactions will be encountered in future
discussions, and five simple examples are shown below. The first and
third equations demonstrate the strongly basic nature of these
compounds, which bond rapidly to the weakly acidic protons of water and
methyl alcohol (colored blue). The nucleophilic carbon of these reagents
also bonds readily with electrophiles such as iodine (second equation)
and carbon dioxide (fifth equation). The polarity of the carbon-oxygen
double bonds of CO2
makes the carbon atom electrophilic, shown by the formula in the shaded
box, so the nucleophilic carbon of the Grignard reagent bonds to this
site. As noted above, solutions of these reagents must also be protected
from oxygen, since peroxides are formed (equation 4).
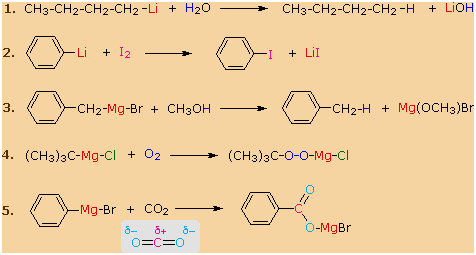
Another important reaction exhibited by these organometallic reagents is
metal exchange. In the first example below, methyl lithium reacts
with cuprous iodide to give a lithium dimethylcopper reagent, which is
referred to as a Gilman reagent. Other alkyl lithiums give
similar Gilman reagents. A useful application of these reagents is their
ability to couple with alkyl, vinyl and aryl iodides, as shown in the
second equation. Later we shall find that Gilman reagents also display
useful carbon-carbon bond forming reactions with conjugated enones and
with acyl chlorides.
|
2 CH3Li
+ CuI
��> (CH3)2CuLi + LiI
Formation of a Gilman Reagent
(C3H7)2CuLi + C6H5I
��>
C6H5-C3H7
+ LiI + C3H7Cu A
Coupling Reaction |
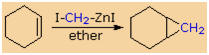
The formation of organometallic reagents from alkyl halides is more
tolerant of structural variation than were the nucleophilic
substitutions described earlier. Changes in carbon hybridization have little effect on the
reaction, and 1�, 2� and 3�-alkyl halides all react in the same manner.
One restriction, of course, is the necessary absence of incompatible
functional groups elsewhere in the reactant molecule. For example,
5-bromo-1-pentanol fails to give a Grignard reagent (or a lithium
reagent) because the hydroxyl group protonates this reactive function as
soon as it is formed.
BrCH2CH2CH2CH2CH2OH
+
Mg
��>
[
BrMgCH2CH2CH2CH2CH2OH
]
��>
HCH2CH2CH2CH2CH2OMgBr
Reactions of Dihalides
If two halogen atoms are present in a given compound, reactions
with reducing metals may take different paths depending on how close the
carbon-halogen bonds are to each other. If they are separated by four or
more carbons, as in the first example below, a bis-organometallic
compound may be formed. However, if the halogens are bonded to adjacent
(vicinal)
carbons, an elimination takes place with formation of a double bond.
Since vicinal-dihalides are usually made by adding a halogen to a double
bond, this reaction is mainly useful for relating structures to each
other. The last example, in which two halogens are bonded to the same
carbon, referred to as geminal (twinned), gives an unusual
reagent which may either react as a carbon nucleophile or, by
elimination, as a
carbene. Such reagents are often termed carbenoid.
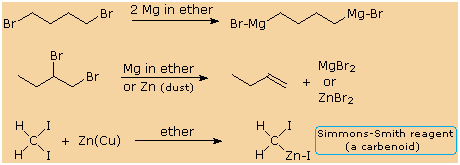
The solution structure of the Simmons-Smith reagent is less well
understood than that of the Grignard reagent, but the formula given here
is as useful as any that have been proposed. Other alpha-halogenated
organometallic reagents, such as ClCH2Li,
BrCH2Li,
Cl2CHLi
and Cl3CLi,
have been prepared, but they are substantially less stable and must be
maintained at very low temperature (ca. -100 � C) to avoid loss of LiX.
The stability and usefulness of the Simmons-Smith reagent may be
attributed in part to the higher covalency of the carbon-zinc bond
together with solvation and internal coordination of the zinc.
Hydrolysis (reaction with water) gives methyl iodide, confirming the
basicity of the carbon; and reaction with alkenes gives cyclopropane
derivatives, demonstrating the carbene-like nature of the reagent. The
latter transformation is illustrated by the equation on the right.
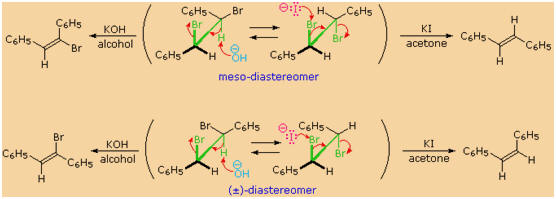
Elimination reactions of the stereoisomeric
1,2-dibromo-1,2-diphenylethanes provide a nice summary of the principles
discussed above. The following illustration shows first the meso-diastereomer
and below it one enantiomer of the racemic-diastereomer. In each case
two conformers are drawn within parentheses, and the anti-relationship
of selected vicinal groups in each is colored green. The reaction
proceeding to the left is a dehydrohalogenation induced by treatment
with KOH in alcohol. Since this is a
stereospecific elimination, each diastereomer gives a
different stereoisomeric product. The reaction to the right is a
dehalogenation (the reverse of halogen addition to an alkene), caused by
treatment with iodide anion. Zinc dust effects the same reaction, but
with a lower degree of stereospecificity. The mechanism of the iodide
anion reaction is shown by red arrows in the top example. A similar
mechanism explains the comparable elimination of the racemic isomer. In
both reactions an anti-transition state is observed.
The two stereoisomers of 1-bromo-1,2-diphenylethene (shown on the
left of the diagram) undergo a second dehydrobromination reaction on
more vigorous treatment with base, as shown in the following equation.
This elimination generates the same alkyne (carbon-carbon triple bond)
from each of the bromo-alkenes. Interestingly, the (Z)-isomer (lower
structure) eliminates more rapidly than the (E)-isomer (upper
structure), again showing a preference for anti-orientation of
eliminating groups.
C6H5CH=CBrC6H5
+ KOH
��>
C6H5C≡CC6H5
+ KBr + H2O
Preparation of Alkynes by
Dehydrohalogenation
The last reaction shown above suggests that alkynes might be
prepared from alkenes by a two stage procedure, consisting first of
chlorine or bromine addition to the double bond, and secondly a base
induced double dehydrohalogenation. For example, reaction of 1-butene
with bromine would give 1,2-dibromobutane, and on treatment with base
this vicinal dibromide would be expected to yield 1-bromo-1-butene
followed by a second elimination to 1-butyne.
CH3CH2CH=CH2
+
Br2
��>
CH3CH2CHBr�CH2Br
+ base
��>
CH3CH2CH=CHBr
+ base
��>
CH3CH2C≡CH
In practice this strategy works, but it requires care in the
selection of the base and solvent. If KOH in alcohol is used, the first
elimination is much faster than the second, so the bromoalkene may be
isolated if desired. Under more extreme conditions the second
elimination takes place, but isomerization of the triple bond also
occurs, with the more stable isomer (2-butyne) being formed along with
1-butyne, even becoming the chief product. To facilitate the second
elimination and avoid isomerization the very strong base sodium amide,
NaNH2,
may be used. Since ammonia is a much weaker acid than water (by a factor
of 1018),
its conjugate base is proportionally stronger than hydroxide anion (the
conjugate base of water), and the elimination of HBr from the
bromoalkene may be conducted at relatively low temperature. Also, the
acidity of the sp-hybridized C-H bond of the terminal alkyne traps the
initially formed 1-butyne in the form of its sodium salt.
CH3CH2C≡CH
+ NaNH2
��>
CH3CH2C≡C:(�)
Na(+)
+ NH3
An additional complication of this procedure is that the
1-bromo-1-butene product of the first elimination (see previous
equations) is accompanied by its 2-bromo-1-butene isomer, CH3CH2CBr=CH2,
and elimination of HBr from this bromoalkene not only gives 1-butyne
(base attack at C-1) but also 1,2-butadiene, CH3CH=C=CH2,
by base attack at C-3. Dienes of this kind, in which the central carbon
is sp-hybridized, are called allenes and are said to have
cumulated double bonds. They are usually less stable than their
alkyne isomers.
Elimination Reactions
1. The E2 Reaction
We
have not yet considered the factors that influence elimination
reactions, such as example
3 in the group presented at the
beginning of this section.
(3)
(CH3)3C-Br
+ CN(�)
��>
(CH3)2C=CH2
+
Br(�)
+ HCN
We know that t-butyl bromide is not expected to react by a SN2
mechanism. Furthermore, the ethanol solvent is not sufficiently polar to
facilitate a SN1
reaction. The other reactant, cyanide anion, is a good nucleophile; and
it is also a decent base, being about ten times weaker than bicarbonate.
Consequently, a base-induced elimination seems to be the only plausible
reaction remaining for this combination of reactants. To get a clearer
picture of the interplay of these factors consider the reaction of a
2�-alkyl halide, isopropyl bromide, with two different nucleophiles.
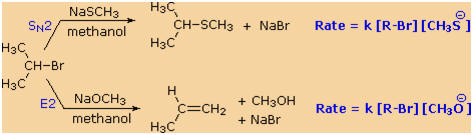
In the methanol solvent used here, methanethiolate has greater
nucleophilicity than methoxide by a factor of 100. Methoxide, on the
other hand is roughly 106
times more basic than methanethiolate. As a result, we see a clearcut
difference in the reaction products, which reflects nucleophilicity
(bonding to an electrophilic carbon) versus basicity (bonding to
a proton). Kinetic studies of these reactions show that they are both
second order (first order in R�Br and first order in Nu:(�)),
suggesting a bimolecular mechanism for each. The substitution reaction
is clearly SN2.
The corresponding designation for the elimination reaction is E2.
An energy diagram for the single-step bimolecular E2 mechanism is shown
on the right. We should be aware that the E2 transition state is less
well defined than is that of SN2 reactions. More bonds are being broken and formed, with the
possibility of a continuum of states in which the extent of C�H and C�X
bond-breaking and C=C bond-making varies. For example, if the R�groups
on the beta-carbon enhance the acidity of that hydrogen, then
substantial breaking of C�H may occur before the other bonds begin to be
affected. Similarly, groups that favor ionization of the halogen may
generate a transition state with substantial positive charge on the
alpha-carbon and only a small degree of C�H breaking. For most simple
alkyl halides, however, it is proper to envision a balanced transition
state, in which there has been an equal and synchronous change in all
the bonds. Such a model helps to explain an important regioselectivity
displayed by these elimination reactions.
If two or more structurally distinct groups of beta-hydrogens are
present in a given reactant, then several constitutionally isomeric
alkenes may be formed by an E2 elimination. This situation is
illustrated by the 2-bromobutane and 2-bromo-2,3-dimethylbutane
elimination examples given below.
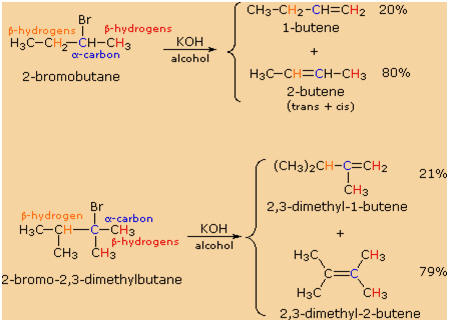
By using the strongly basic hydroxide nucleophile, we direct these
reactions toward elimination. In both cases there are two different sets
of beta-hydrogens available to the elimination reaction (these are
colored red and orange and the alpha carbon is blue). If the rate of
each possible elimination was the same, we might expect the amounts of
the isomeric elimination products to reflect the number of hydrogens
that could participate in that reaction. For example, since there are
three 1�-hydrogens (red) and two 2�-hydrogens (orange) on beta-carbons
in 2-bromobutane, statistics would suggest a 3:2 ratio of 1-butene and
2-butene in the products. This is not observed, and the latter
predominates by 4:1. This departure from statistical expectation is even
more pronounced in the second example, where there are six
1�-beta-hydrogens compared with one 3�-hydrogen. These results point to
a strong regioselectivity favoring the more highly substituted product
double bond, an empirical statement generally called the Zaitsev Rule.
The main factor contributing to Zaitsev Rule behavior is the
stability of the alkene. We noted earlier that carbon-carbon double
bonds are stabilized (thermodynamically) by alkyl substituents, and that
this stabilization could be evaluated by appropriate
heat of hydrogenation measurements. Since the E2 transition
state has significant carbon-carbon double bond character, alkene
stability differences will be reflected in the transition states of
elimination reactions, and therefore in the activation energy of the
rate-determining steps. From this consideration we anticipate that if
two or more alkenes may be generated by an E2 elimination, the more
stable alkene will be formed more rapidly and will therefore be the
predominant product. This is illustrated for 2-bromobutane by the energy
diagram on the right. The propensity of E2 eliminations to give the more
stable alkene product also influences the distribution of product
stereoisomers. In the elimination of 2-bromobutane, for example, we find
that trans-2-butene is produced in a 6:1 ratio with its cis-isomer.
The Zaitsev Rule is a good predictor for simple elimination reactions of
alkyl chlorides, bromides and iodides as long as relatively small strong
bases are used. Thus hydroxide, methoxide and ethoxide bases give
comparable results. Bulky bases such as tert-butoxide tend to give
higher yields of the less substituted double bond isomers, a
characteristic that has been attributed to steric hindrance. In the case
of 2-bromo-2,3-dimethylbutane, described above, tert-butoxide gave a 4:1
ratio of 2,3-dimethyl-1-butene to 2,3-dimethyl-2-butene ( essentially
the opposite result to that obtained with hydroxide or methoxide). This
point will be discussed further once we know more about the the
structure of the E2 transition state.
Bredt's Rule
The importance of maintaining a planar configuration of the trigonal
double-bond carbon components must never be overlooked. For optimum
pi-bonding to occur, the p-orbitals on these carbons must be parallel,
and the resulting doubly-bonded planar configuration is more stable than
a twisted alternative by over 60 kcal/mole. This structural constraint
is responsible for the existence of
alkene stereoisomers when substitutuion patterns permit. It
also prohibits certain elimination reactions of bicyclic alkyl halides,
that might be favorable in simpler cases. For example, the bicyclooctyl
3�-chloride shown below appears to be similar to tert-butyl chloride,
but it does not undergo elimination, even when treated with a strong
base (e.g. KOH or KOC4H9).
There are six equivalent beta-hydrogens that might be attacked by base
(two of these are colored blue as a reference), so an E2 reaction seems
plausible. The problem with this elimination is that the resulting
double bond would be constrained in a severely twisted (non-planar)
configuration by the bridged structure of the carbon skeleton. The
carbon atoms of this twisted double-bond are colored red and blue
respectively, and a Newman projection looking down the twisted bond is
drawn on the right. Because a pi-bond cannot be formed, the hypothetical
alkene does not exist. Structural prohibitions such as this are often
encountered in small bridged ring systems, and are referred to as
Bredt's Rule.
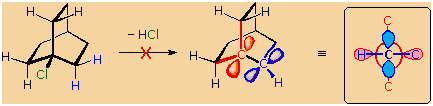
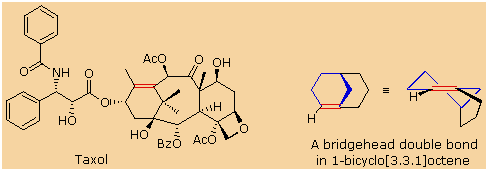
Bredt's Rule should not be applied blindly to all bridged ring systems.
If large rings are present their conformational flexibility may permit
good overlap of the p-orbitals of a double bond at a bridgehead. This is
similar to recognizing that trans-cycloalkenes cannot be prepared if the
ring is small (3 to 7-membered), but can be isolated for larger ring
systems. The anti-tumor agent taxol has such a bridgehead double bond
(colored red), as shown in the following illustration. The
bicyclo[3.3.1]octane ring system is the smallest in which bridgehead
double bonds have been observed. The drawing to the right of taxol shows
this system. The bridgehead double bond (red) has a cis-orientation in
the six-membered ring (colored blue), but a trans-orientation in the
larger eight-membered ring.
2. Stereochemistry of the E2
Reaction
E2 elimination reactions of certain isomeric cycloalkyl halides show
unusual rates and regioselectivity that are not explained by the
principles thus far discussed. For example,
trans-2-methyl-1-chlorocyclohexane reacts with alcoholic KOH at a much
slower rate than does its cis-isomer. Furthermore, the product from
elimination of the trans-isomer is 3-methylcyclohexene (not predicted by
the Zaitsev rule), whereas the cis-isomer gives the predicted
1-methylcyclohexene as the chief product. These differences are
described by the first two equations in the following diagram.
Unlike open chain structures, cyclic compounds generally restrict the
spatial orientation of ring substituents to relatively few arrangements.
Consequently, reactions conducted on such substrates often provide us
with information about the preferred orientation of reactant species in
the transition state. Stereoisomers are particularly suitable in this
respect, so the results shown here contain important information about
the E2 transition state.
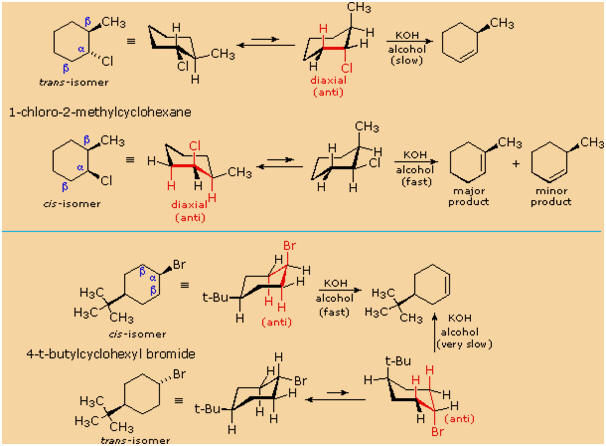
The most sensible interpretation of the elimination reactions of 2- and
4-substituted halocyclohexanes is that this reaction prefers an anti
orientation of the halogen and the beta-hydrogen which is attacked
by the base. These anti orientations are colored in red in the above
equations. The compounds used here all have six-membered rings, so the
anti orientation of groups requires that they assume a diaxial
conformation. The observed differences in rate are the result of a
steric preference for
equatorial orientation of large substituents,
which reduces the effective concentration of conformers having an axial
halogen. In the case of the 1-bromo-4-tert-butylcyclohexane isomers, the
tert-butyl group is so large that it will always assume an equatorial
orientation, leaving the bromine to be axial in the cis-isomer and
equatorial in the trans. Because of symmetry, the two axial
beta-hydrogens in the cis-isomer react equally with base, resulting in
rapid elimination to the same alkene (actually a racemic mixture). This
reflects the fixed anti orientation of these hydrogens to the chlorine
atom. To assume a conformation having an axial bromine the trans-isomer
must tolerate serious crowding distortions. Such conformers are
therefore present in extremely low concentration, and the rate of
elimination is very slow. Indeed, substitution by hydroxide anion
predominates.
A similar analysis of the 1-chloro-2-methylcyclohexane isomers explains
both the rate and regioselectivity differences. Both the chlorine and
methyl groups may assume an equatorial orientation in a chair
conformation of the trans-isomer, as shown in the top equation. The
axial chlorine needed for the E2 elimination is present only in the less
stable alternative chair conformer, but this structure has only one
axial beta-hydrogen (colored red), and the resulting elimination gives
3-methylcyclohexene. In the cis-isomer the smaller chlorine atom assumes
an axial position in the more stable chair conformation, and here there
are two axial beta hydrogens. The more stable 1-methylcyclohexene is
therefore the predominant product, and the overall rate of elimination
is relatively fast.
An orbital drawing of the anti-transition state is shown on the right.
Note that the base attacks the alkyl halide from the side opposite the
halogen, just as in the SN2
mechanism. In this drawing the α and β carbon atoms are undergoing a
rehybridization from sp3
to sp2
and the developing π-bond is drawn as dashed light blue lines. The
symbol R represents an alkyl group or hydrogen. Since both the
base and the alkyl halide are present in this transition state, the
reaction is bimolecular and should exhibit second order kinetics. We
should note in passing that a syn-transition state would also provide
good orbital overlap for elimination, and in some cases where an
anti-orientation is prohibited by structural constraints syn-elimination
has been observed.
It is also worth noting that anti-transition states were preferred in
several addition reactions to alkenes, so there is an intriguing
symmetry to these inverse structural transformations.Having arrived at a
useful and plausible model of the E2 transition state, we can understand
why a bulky base might shift the regioselectivity of the reaction away
from the most highly substituted double bond isomer. Steric hindrance to
base attack at a highly substituted beta-hydrogen site would result in
preferred attack at a less substituted site.
To see the effect of steric hindrance at a beta carbon on the E2
transition state
3. The E1 Reaction
Just as there were two mechanisms for nucleophilic substitution, there
are two elimination mechanisms. The E1 mechanism is nearly identical to
the SN1
mechanism, differing only in the course of reaction taken by the
carbocation intermediate. As shown by the following equations, a
carbocation bearing beta-hydrogens may function either as a Lewis acid
(electrophile), as it does in the SN1
reaction, or a Br�nsted acid, as in the E1 reaction.
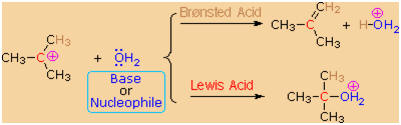
Thus, hydrolysis of tert-butyl chloride in a mixed solvent of water and
acetonitrile gives a mixture of 2-methyl-2-propanol (60%) and
2-methylpropene (40%) at a rate independent of the water concentration.
The alcohol is the product of an SN1
reaction and the alkene is the product of the E1 reaction. The
characteristics of these two reaction mechanisms are similar, as
expected. They both show first order kinetics; neither is much
influenced by a change in the nucleophile/base; and both are relatively
non-stereospecific.
(CH3)3C�Cl
+ H2O
��>
[ (CH3)3C(+)
] +
Cl(�)
+ H2O
��>
(CH3)3C�OH
+ (CH3)2C=CH2
+ HCl + H2O
To summarize, when carbocation intermediates are formed one can expect
them to react further by one or more of the following modes:
1.
The cation may bond to a nucleophile to give a substitution product.
2. The cation may transfer a beta-proton to a base, giving an
alkene product.
3. The cation may rearrange to a more stable carbocation, and
then react by mode #1 or #2.
Since the SN1
and E1 reactions proceed via the same carbocation intermediate, the
product ratios are difficult to control and both substitution and
elimination usually take place.
Having discussed the many factors that influence nucleophilic
substitution and elimination reactions of alkyl halides, we must now
consider the practical problem of predicting the most likely outcome
when a given alkyl halide is reacted with a given nucleophile. As we
noted earlier, several variables must be considered, the most
important being the structure of the alkyl group and the nature of the
nucleophilic reactant. The nature of the halogen substituent on the
alkyl halide is usually not very significant if it is Cl, Br or I. In
cases where both SN2 and E2 reactions compete, chlorides generally give more
elimination than do iodides, since the greater electronegativity of
chlorine increases the acidity of beta-hydrogens. Indeed, although alkyl
fluorides are relatively unreactive, when reactions with basic
nucleophiles are forced, elimination occurs (note the high
electronegativity of fluorine).
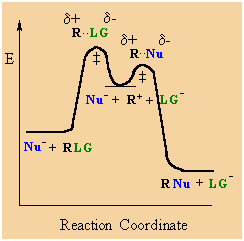
SN1 mechanism
SN1
indicates a substitution, nucleophilic, unimolecular reaction,
described by the expression rate = k [R-LG]
This
pathway is a multi-step process with the following characteristics:
-
step 1: rate
determining (slow) loss of the leaving group,
LG, to generate a carbocation
intermediate, then
-
step 2: rapid
attack of a nucleophile on the electrophilic carbocation to form a new σbond
|
 |
Multi-step reactions have intermediates and a several transition
states (TS).
In an SN1
there is loss of the leaving group generates an intermediate
carbocation which is then undergoes a rapid reaction with the
nucleophile.
The reaction profiles
shown here are simplified and do not include the equilibria for
protonation of the -OH. |
 |
|
General case |
|
SN1 reaction |
The
following issues are relevant to the SN1 reactions of alcohols:
Effect of R-
Reactivity order : (CH3)3C- > (CH3)2CH-
> CH3CH2- > CH3-
In an
SN1 reaction, the key step is the loss of the leaving group to form
the intermediate carbocation. The more stable the carbocation is, the easier it
is to form, and the faster the SN1 reaction will be. Some students
fall into the trap of thinking that the system with the less stable carbocation
will react fastest, but they are forgetting that it is the generation of the
carbocation that is rate determining.
-LG
The only event in the rate determining step of the SN1 is breaking
the C-LG bond. For alcohols it
is important to remember that -OH is a very poor leaving. In the reactions with
HX, the -OH is protonated first to give an oxonium, providing the much better
leaving group, a water molecule (see scheme below).
Nu
Since the nucleophile is not involved in the rate determining step of an SN1
reaction, the nature of the nucleophile is unimportant. In the reactions of
alcohols with HX, the reactivity trend of HI > HBr > HCl > HF is not due to the
nucleophilicity of the halide ion but the acidity of HX which is involved in
generating the leaving group prior to the rate determining step.
Stereochemistry
|
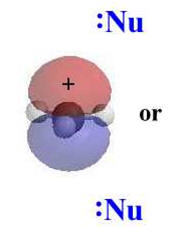 |
In an SN1, the nucleophile attacks the planar carbocation.
Since there is an equally probability of attack on either face there
will be a loss of stereochemistry at the reactive center and
both possible products will be observed.
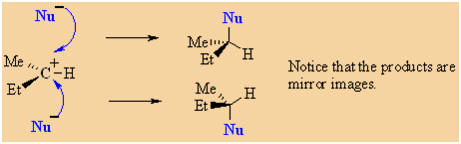 |
Carbocations
Stability:
The general stability order of simple alkyl carbocations is: (most stable) 3o
> 2o > 1o > methyl (least stable)
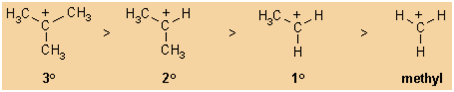
This is because alkyl groups are weakly electron donating due to
hyperconjugation and
inductive effects. Resonance effects can further stabilise carbocations
when present (delocalisation of charge is a stabilising effect).
Note that reactions that occur via 3o and 2o are
known. 1o cations have been observed under special conditions,
but methyl cations have never been observed.
Structure:
|
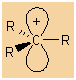 |
Alkyl carbocations are sp2 hybridised, planar systems at
the cationic C centre.
The p-orbital that is not utilised in the hybrids is empty and is
often shown bearing the positive charge since it represents the
orbital available to accept electrons. |
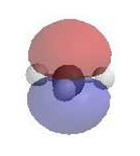 |
Reactivity:
|
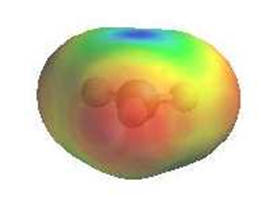 |
As they have an incomplete octet, carbocations are excellent
electrophiles and react readily with nucleophiles.
Alternatively, loss of H+ can generate a π bond.
The electrostatic
potential diagrams clearly show the cationic center in
blue, this is where the
nucleophile will attack.
|
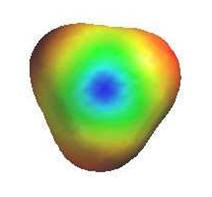 |
Rearrangements:
Carbocations are prone to rearrangement via 1,2-hydride or 1,2-alkyl shifts if
it generates a more stable carbocation.
Hyper conjugation
Hyperconjugation
is the stabilising interaction that results from the interaction of the
electrons in a σ-bond (usually C-H or C-C) with an adjacent empty
(or partially filled) p-orbital or a π-orbital to give an extended molecular
orbital that increases the stability of the system.
Based on the
valence bond model of bonding, hyperconjugation can be described as "double bond
- no bond resonance" but it is not what we would "normally" call resonance,
though the similarity is shown below.
Hyperconjugation
is a factor in explaining why increasing the number of alkyl substituents on a
carbocation or radical centre leads to an increase in stability.
|
Let's
consider how a methyl group is involved in hyperconjugation with a
carbocation centre. |
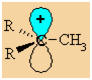 |
|
First
we need to draw it to show the C-H σ-bonds.
Note that the empty p orbital associated with the positive charge at
the carbocation centre is in the same plane (i.e. coplanar)
with one of the C-H
σ-bonds (shown in blue.) |
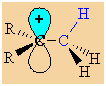 |
|
This
geometry means the electrons in the σ-bond can be stabilised by an
interaction with the empty p-orbital of the carbocation centre.
(this
diagram shows the similarity with resonance and the structure on the
right has the "double bond - no bond" character) |
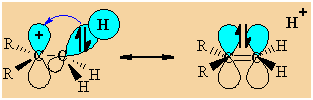 |
-
The
stabilisation arises because the orbital interaction leads to the electrons
being in a lower energy orbital
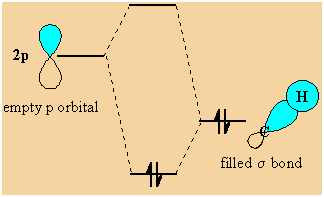
-
Of course,
the C-C σ-bond is free to rotate, and as it does so, each of the
C-H σ-bonds in turn undergoes the stabilising interaction.
-
The ethyl
cation has 3 C-H σ-bonds that can be involved in hyperconjugation.
-
The more
hyperconjuagtion there is, the greater the stabilisation of the system.
-
For example,
the t-butyl cation has 9 C-H σ-bonds that can be involved in
hyperconjugation.
-
Hence (CH3)3C+
is more stable than CH3CH2+
-
The effect is
not limited to C-H σ-bonds, appropriate C-C σ-bonds can also
be involved in hyperconjugation.
Inductive effects
-
An
inductive effect is an electronic effect due to the polarisation of σ
bonds within a molecule or ion.
-
In a
carbocation, the positive C attracts the electrons in the σ bonds towards
itself and away from the atom at the other end of the s bond.
-
Electrons in
C-C bonds are more readily polarised than those in a C-H bond.
-
Therefore,
alkyl groups are better at stabilising C+ than H atoms.
Go to Main Menu
-
© M.EL-Fellah ,Chemistry
Department, Garyounis University
|